Co-reporter:Kai C. Göbgen, Simon Steinberg, and Richard Dronskowski
Inorganic Chemistry September 18, 2017 Volume 56(Issue 18) pp:11398-11398
Publication Date(Web):August 28, 2017
DOI:10.1021/acs.inorgchem.7b01847
Through explorations of the silicon–tellurium system we identified the extremely air-sensitive, red Si1.67(4)Te3≡Si1.11(3)Te2 that is a silicon-deficient relative of the previously reported Si2Te3. The crystal structure features hexagonal closest packed layers of tellurium atoms with disordered [Si2] dumbbells residing in about 50% of the octahedra of every second layer enclosed by the tellurium atoms. In addition to the determination of the crystal structure for this silicon telluride, we probed the opportunity of the existence of a SiTe2 adopting the Si2Te3-structure by means of quantum chemical techniques. The investigations of the electronic structures and a subsequent chemical bonding analysis based on the projected Crystal Orbital Hamilton Population (pCOHP) technique for two “SiTe2” models revealed a tendency to align the [Si2] dumbbells parallel to the c axis to maximize Si–Te bonding. However, the disorder of the [Si2] dumbbells appears to be a consequence of non-equilibrium condensation into the solid state.
Co-reporter:T. Scholz;R. Dronskowski
Journal of Materials Chemistry C 2017 vol. 5(Issue 1) pp:166-175
Publication Date(Web):2016/12/22
DOI:10.1039/C6TC04543J
We present an experimental and theoretical study of the solid solution GexFe4−xNy (0 ≤ x ≤ 1). A two-step ammonolytic reaction gives access to the compounds with phase-pure quality. The GexFe4−xNy nitrides show a transition from an antiperovskite-like to a tetragonally distorted structure with increasing germanium concentration. Various experimental and theoretical methods evidence that the iron substitution by germanium exclusively takes place at the cubic Wyckoff position 1a. Despite the phase transition, one observes a Vegard-type decrease of the lattice parameter over the entire compositional range. In addition, the nitrides have a limited nitrogen capacity: incorporating germanium drastically reduces the nitrogen content in the cubic structure, but increases it again in the tetragonal structure. Combined HT-XRD and TG-DSC measurements evidence that the germanium-richest nitrides are highly expanding materials. They first show a transition to a cubic structure before they decompose by releasing nitrogen. Magnetic measurements reveal that the gradual germanium incorporation is accompanied by a drastic weakening of the ferromagnetic interactions leading to a frustrated iron spin system. Ge0.97Fe3.03N0.56 is identified as a canonical spin glass with the characteristic parameters Tg = 36.68(5) K, τ* = 10−13.8(2) s, zν = 7.2(1) and ΔTm/(Tm·Δ lg ω) = 0.012.
Co-reporter:Hannes Dierkes, Jan van Leusen, Dimitri Bogdanovski, and Richard Dronskowski
Inorganic Chemistry 2017 Volume 56(Issue 3) pp:
Publication Date(Web):January 17, 2017
DOI:10.1021/acs.inorgchem.6b02816
κ-carbides of varying composition, seemingly responsible for age hardening in high-Al steel alloys, have been detected to precipitate both at grain boundaries and in the bulk grain of steels. Herein we report the bulk-phase synthesis of “Mn3AlC” by arc plasma sintering and rapid solidification. Single crystals have been found suitable for X-ray diffraction using Mo radiation and yield a lattice parameter of a = 3.875(2) Å. We find a mixed occupation of the 1a position by Al and Mn, which, together with the C position being fully occupied, leads to the actual composition Mn3.1Al0.9C. Additional energy-dispersive X-ray–scanning electron microscopy measurements support the composition and corroborate the homogeneity. SQUID data collected on the polycrystalline ferromagnetic sample exhibit a Curie temperature of about 295 ± 13 K and a soft magnetic behavior. The small but significant nonstoichiometry on 1a leads to a slightly larger lattice parameter, a higher electron count, and, thus, a lowered density of states at the Fermi level, indicative of increased phase stability.
Co-reporter:Janine GeorgeRichard Dronskowski
The Journal of Physical Chemistry A 2017 Volume 121(Issue 6) pp:
Publication Date(Web):January 20, 2017
DOI:10.1021/acs.jpca.6b12732
Intermolecular bonds play a crucial role in the rational design of crystal structures, dubbed crystal engineering. The relatively new term tetrel bonds (TBs) describes a long-known type of such interactions presently in the focus of quantum chemical cluster calculations. Here, we energetically explore the strengths and cooperativity of these interactions in infinite chains, a possible arrangement of such tetrel bonds in extended crystals, by periodic density functional theory. In the chains, the TBs are amplified due to cooperativity by up to 60%. Moreover, we computationally take apart crystals stabilized by infinite tetrel-bonded chains and assess the importance of the TBs for the crystal stabilization. Tetrel bonds can amount to 70% of the overall interaction energy within some crystals, and they can also be energetically decisive for the taken crystal structure; their individual strengths also compete with the collective packing within the crystal structures.
Co-reporter:M. Sc. Michael Küpers;M. Sc. Philipp M. Konze;Dipl.-Chem. Stefan Maintz;Dr. Simon Steinberg;Dr. Antonio M. Mio;Dr. Oana Cojocaru-Mirédin;Dr. Min Zhu;Dr. Merlin Müller;Dr. Martina Luysberg; Dr. Joachim Mayer; Dr. Matthias Wuttig; Dr. Richard Dronskowski
Angewandte Chemie 2017 Volume 129(Issue 34) pp:10381-10381
Publication Date(Web):2017/08/14
DOI:10.1002/ange.201706616
Unerwartete Ge-Ge-Wechselwirkungen im “zweidimensionalen” Ge4Se3Te fanden R. Dronskowski et al., wie in der Zuschrift auf S. 10338 berichtet wird. Die Schichtverbindung wurde mittels chemischen Transports kristallisiert und erstmals strukturell charakterisiert. Ihre elektronische Struktur und der Ursprung der Ge-Ge-Wechselwirkungen wurden mithilfe chemischer Bindungsanalyse und der neu eingeführten Energiedichte(DOE)-Funktion beleuchtet.
Co-reporter:M. Sc. Michael Küpers;M. Sc. Philipp M. Konze;Dipl.-Chem. Stefan Maintz;Dr. Simon Steinberg;Dr. Antonio M. Mio;Dr. Oana Cojocaru-Mirédin;Dr. Min Zhu;Dr. Merlin Müller;Dr. Martina Luysberg; Dr. Joachim Mayer; Dr. Matthias Wuttig; Dr. Richard Dronskowski
Angewandte Chemie International Edition 2017 Volume 56(Issue 34) pp:10204-10208
Publication Date(Web):2017/08/14
DOI:10.1002/anie.201612121
AbstractA hexagonal phase in the ternary Ge–Se–Te system with an approximate composition of GeSe0.75Te0.25 has been known since the 1960s but its structure has remained unknown. We have succeeded in growing single crystals by chemical transport as a prerequisite to solve and refine the Ge4Se3Te structure. It consists of layers that are held together by van der Waals type weak chalcogenide–chalcogenide interactions but also display unexpected Ge–Ge contacts, as confirmed by electron microscopy analysis. The nature of the electronic structure of Ge4Se3Te was characterized by chemical bonding analysis, in particular by the newly introduced density of energy (DOE) function. The Ge–Ge bonding interactions serve to hold electrons that would otherwise go into antibonding Ge–Te contacts.
Co-reporter:M. Sc. Michael Küpers;M. Sc. Philipp M. Konze;Dipl.-Chem. Stefan Maintz;Dr. Simon Steinberg;Dr. Antonio M. Mio;Dr. Oana Cojocaru-Mirédin;Dr. Min Zhu;Dr. Merlin Müller;Dr. Martina Luysberg; Dr. Joachim Mayer; Dr. Matthias Wuttig; Dr. Richard Dronskowski
Angewandte Chemie 2017 Volume 129(Issue 34) pp:10338-10342
Publication Date(Web):2017/08/14
DOI:10.1002/ange.201612121
AbstractIm ternären System Ge-Se-Te ist seit den 1960er Jahren eine hexagonale Phase mit der annähernden Zusammensetzung GeSe0.75Te0.25 bekannt, aber ihre Struktur blieb ungeklärt. Durch chemischen Transport gelang nun die Zucht von Einkristallen zur Bestimmung der Ge4Se3Te-Struktur. Diese besteht aus Schichten, die – wie elektronenmikroskopische Analysen erhärten – über van-der-Waals-artige schwache Chalkogen-Chalkogen-, aber auch über unerwartete Ge-Ge-Wechselwirkungen verbunden sind. Die Art der elektronischen Struktur von Ge4Se3Te wurde durch Analyse der chemischen Bindungen bestimmt, speziell mithilfe der neu eingeführten Energiedichte(DOE)-Funktion. Die bindenden Ge-Ge-Wechselwirkungen dienen der Aufnahme von Elektronen, die ansonsten antibindende Ge-Te-Zustände einnähmen.
Co-reporter:Marc Esser, Arina A. Esser, Davide M. Proserpio, Richard Dronskowski
Carbon 2017 Volume 121(Volume 121) pp:
Publication Date(Web):1 September 2017
DOI:10.1016/j.carbon.2017.05.062
Utilizing first-principles electronic-structure calculations, we present the chemical-bonding analyses of hypothetical carbon allotropes based on tetrahedral structure motifs such as T-carbon, TY-carbon and T-graphene. While previous publications on these novel allotropes have dealt with ab initio phonon, band structure and DOS calculations, the focus of this work is the partitioning of the band-structure energy in terms of bonding, nonbonding and antibonding contributions. We re-evaluate the chance of making such allotropes by careful bond analyses and compare them to already known equivalents, namely diamond, graphene and the Buckminsterfullerene molecule. A synthetic route is proposed to a new compound, called TY-carbodiimide, that exhibits similar structure and bonding properties as TY-carbon.Download high-res image (253KB)Download full-size image
Co-reporter:M. Sc. Michael Küpers;M. Sc. Philipp M. Konze;Dipl.-Chem. Stefan Maintz;Dr. Simon Steinberg;Dr. Antonio M. Mio;Dr. Oana Cojocaru-Mirédin;Dr. Min Zhu;Dr. Merlin Müller;Dr. Martina Luysberg; Dr. Joachim Mayer; Dr. Matthias Wuttig; Dr. Richard Dronskowski
Angewandte Chemie International Edition 2017 Volume 56(Issue 34) pp:10247-10247
Publication Date(Web):2017/08/14
DOI:10.1002/anie.201706616
Unexpected Ge–Ge interactions were found in “two-dimensional” Ge4Se3Te as reported by R. Dronskowski et al. in their Communication on page 10204 ff. The layered material was crystallized using chemical vapor deposition and then characterized for the first time. Its electronic structure and the chemical cause of the Ge–Ge interactions were examined by chemical bonding analysis and the newly introduced density of energy (DOE) function.
Co-reporter:Marc Esser, Richard Dronskowski
Carbon 2017 Volume 123(Volume 123) pp:
Publication Date(Web):1 October 2017
DOI:10.1016/j.carbon.2017.08.010
Recent years have brought forth an ever-increasing number of predicted carbon allotropes. In the spirit of Heimann's original categorization scheme for carbon-only materials, we here report an ab initio method to create triangular (ternary) maps based on their valence-orbital mixing. These maps group together allotropes of similar electronic structure––and, hence, physical properties––and can thus aid in finding allotropes with specific features. Moreover, these maps can be used to classify all carbon allotropes according to their bonding nature. We suggest to extract insights about the composition of the crystal wave function as emerging from individual atomic orbitals. To do so, we develop a way to visualize the entire linear-coefficient space of an extended LCAO wave function, also based on ternary diagrams. This scheme yields that lower-level mixed states always get realized and filled first before higher-level mixed states can be created, a consequence of symmetry breaking of canonical orbitals within the solid state. For the specific case of graphite, there is more sp mixing than in sp2 mixing present, while diamond exhibits more sp and sp2 mixing than sp3 mixing. Once higher-level mixing is realized, however, these levels are more significant for the allotropes' properties than the lower-level ones.Download high-res image (161KB)Download full-size image
Co-reporter:Philipp M. Konze, Volker L. Deringer, and Richard Dronskowski
Chemistry of Materials 2016 Volume 28(Issue 18) pp:6682
Publication Date(Web):August 25, 2016
DOI:10.1021/acs.chemmater.6b02940
Nanocrystals with rationally tailored morphologies and properties are a thriving research field. The phase-change material (PCM) germanium telluride (GeTe) has recently been synthesized in a plethora of nanocrystalline forms and morphologies, but a mechanistic understanding of these experimental findings has been largely missing so far. Here, we present comprehensive dispersion-corrected density functional theory (DFT-D) simulations of the low-index crystal facets of GeTe and how they interact with relevant ligand molecules. These results enable a systematic understanding of experimental findings regarding GeTe nanocrystals and clarify the origin of the broad range of particle shapes observed. This study also affords semiquantitative signposts assisting future synthetic studies of nanocrystalline PCMs.
Co-reporter:Arno L. Görne, Janine George, Jan van Leusen, Gerald Dück, Philipp Jacobs, Naveen Kumar Chogondahalli Muniraju, and Richard Dronskowski
Inorganic Chemistry 2016 Volume 55(Issue 12) pp:6161-6168
Publication Date(Web):June 1, 2016
DOI:10.1021/acs.inorgchem.6b00736
We report the oxidation-controlled synthesis of the ytterbium amides Yb(NH2)2 and Yb(NH2)3 and the first rare-earth-metal guanidinates YbC(NH)3 and Yb(CN3H4)3 from liquid ammonia. For Yb(NH2)2, we present experimental atomic displacement parameters from powder X-ray diffraction (PXRD) and density functional theory (DFT)-derived hydrogen positions for the first time. For Yb(NH2)3, the indexing proposal based on PXRD arrives at R3̅, a = 6.2477(2) Å, c = 17.132(1) Å, V = 579.15(4) Å3, and Z = 6. The oxidation-controlled synthesis was also applied to make the first rare-earth guanidinates, namely, the doubly deprotonated YbC(NH)3 and the singly deprotonated Yb(CN3H4)3. YbC(NH)3 is isostructural with SrC(NH)3, as derived from PXRD (P63/m, a = 5.2596(2) Å, c = 6.6704(2) Å, V = 159.81(1) Å3, and Z = 2). Yb(CN3H4)3 crystallizes in a structure derived from the [ReO3] type, as studied by powder neutron diffraction (Pn3̅, a = 13.5307(3) Å, V = 2477.22(8) Å3, and Z = 8 at 10 K). Electrostatic and hydrogen-bonding interactions cooperate to stabilize the structure with wide and empty channels. The IR spectra of the guanidinates are compared with DFT-calculated phonon spectra to identify the vibrational modes. SQUID magnetometry shows that Yb(CN3H4)3 is a paramagnet with isolated Yb3+ (4f13) ions. A CONDON 2.0 fit was used to extract all relevant parameters.
Co-reporter:Simon Steinberg, Ralf P. Stoffel, and Richard Dronskowski
Crystal Growth & Design 2016 Volume 16(Issue 11) pp:6152
Publication Date(Web):October 12, 2016
DOI:10.1021/acs.cgd.6b01163
A1B1-type tellurides of group 14 elements are of great interest due to their applications as data and energy storage materials. While the features of ATe (A = Ge, Sn, Pb) have been determined, there is no report on SiTe in the solid state. Herein, we review a preexisting controversy in the literature regarding the Si–Te system and provide a feasible approach to SiTe.
Co-reporter:Volker L. Deringer, Ulli Englert, and Richard Dronskowski
Biomacromolecules 2016 Volume 17(Issue 3) pp:
Publication Date(Web):February 1, 2016
DOI:10.1021/acs.biomac.5b01653
Chitin is an abundant biopolymer that stabilizes the exoskeleton of insects and gives structure to plants. Its macroscopic properties go back to an intricate network of hydrogen bonds that connect the polymer strands, and these intermolecular links have been under ongoing study. Here, we use atomistic simulations to explore hydrogen bonding in the most abundant form, α-chitin. The crystal structure exhibits disorder, and so discrete models are systematically derived as suitable approximants to the macroscopic material. These models then allow us to perform dispersion-corrected density-functional theory (DFT-D) simulations on the three-dimensional crystal network and on lower-dimensional fragments. Thereby, we rationalize the nature of hydrogen bonding and the role of crystallographic disorder for the stability of α-chitin, and complement previous, larger-scale molecular-dynamics (MD) simulations as well as recent fiber-diffraction experiments. Our results provide new, atomic-level insight into one of Nature’s most abundant building materials, and the techniques and concepts are likely transferable to other biopolymers.
Co-reporter:Dr. Moulay T. Sougrati;Dr. Ali Darwiche;Dr. Xiaohiu Liu;Dr. Abdelfattah Mahmoud;Dr. Raphael P. Hermann;Dr. Samuel Jouen;Dr. Laure Monconduit;Dr. Richard Dronskowski;Dr. Lorenzo Stievano
Angewandte Chemie International Edition 2016 Volume 55( Issue 16) pp:5090-5095
Publication Date(Web):
DOI:10.1002/anie.201600098
Abstract
We report evidence for the electrochemical activity of transition-metal carbodiimides versus lithium and sodium. In particular, iron carbodiimide, FeNCN, can be efficiently used as negative electrode material for alkali-metal-ion batteries, similar to its oxide analogue FeO. Based on 57Fe Mössbauer and infrared spectroscopy (IR) data, the electrochemical reaction mechanism can be explained by the reversible transformation of the Fe−NCN into Li/Na−NCN bonds during discharge and charge. These new electrode materials exhibit higher capacity compared to well-established negative electrode references such as graphite or hard carbon. Contrary to its oxide analogue, iron carbodiimide does not require heavy treatments (such as nanoscale tailoring, sophisticated textures, or coating) to obtain long cycle life with current density as high as 9 A g−1 for hundreds of charge–discharge cycles. Similar to the iron compound, several other transition-metal carbodiimides Mx(NCN)y with M=Mn, Cr, Zn can cycle successfully versus lithium and sodium. Their electrochemical activity and performance open the way to the design of a novel family of anode materials.
Co-reporter:Dr. Moulay T. Sougrati;Dr. Ali Darwiche;Dr. Xiaohiu Liu;Dr. Abdelfattah Mahmoud;Dr. Raphael P. Hermann;Dr. Samuel Jouen;Dr. Laure Monconduit;Dr. Richard Dronskowski;Dr. Lorenzo Stievano
Angewandte Chemie 2016 Volume 128( Issue 16) pp:5174-5179
Publication Date(Web):
DOI:10.1002/ange.201600098
Abstract
Wir weisen die elektrochemische Aktivität von Übergangsmetallcarbodiimiden gegenüber Lithium und Natrium nach. Insbesondere das Eisencarbodiimid FeNCN läßt sich effektiv als negatives Elektrodenmaterial für Alkalimetallionenbatterien verwenden, ähnlich dem Oxidanalogon FeO. Auf der Basis von 57Fe-Mößbauer- und infrarotspektroskopischen (IR) Daten kann der elektrochemische Reaktionsmechanismus bei Entladung und Beladung durch die reversible Umwandlung von Fe-NCN- in Li/Na-NCN-Bindungen erklärt werden. Diese neuen Elektrodenmaterialien weisen höhere Kapazitäten als die etablierten negativen Referenzelektroden wie Graphit oder Hartkohlenstoff auf. Im Gegensatz zu seinem Oxidanalogon benötigt Eisencarbodiimid keine aufwendige Vorbehandlung (Nanopräparation, spezielle Texturen, Beschichtung usw.), um eine lange Lebensdauer bei Stromdichten bis zu 9 A g−1 für hunderte von Lade-/Entladezyklen zu erreichen. Ähnlich zur Eisenverbindung können einige andere Übergangsmetallcarbodiimide Mx(NCN)y mit M=Mn, Cr, Zn ebenso erfolgreich gegen Lithium und Natrium zyklisieren. Ihre elektrochemische Aktivität und Leistung öffnet den Weg zum Design einer neuartigen Klasse von Anodenmaterialien.
Co-reporter:Volker L. Deringer
The Journal of Physical Chemistry C 2016 Volume 120(Issue 16) pp:8813-8820
Publication Date(Web):April 12, 2016
DOI:10.1021/acs.jpcc.6b02173
Lead sulfide (PbS) and lead selenide (PbSe) are functional materials with manifold applications. In particular, nanocrystals and other nanostructures of PbS and PbSe are of mounting interest, which calls for a thorough understanding of the underlying crystal surfaces. Here, we present a comprehensive density-functional theory (DFT) survey of structures and stabilities for the (001), (011), and (111) surfaces of rocksalt-type PbS and PbSe. A representative set of possible reconstructions is explored for the polar (111) surfaces, allowing us to suggest both 2 × 1 and “octopolar” 2 × 2 motifs as favorable options; this agrees with previous experiments and could guide future ones. With regard to methodology, we address the role of dispersion interactions for studying PbS and PbSe surfaces: interestingly, the prediction of (001) surface energies depends crucially on the use of dispersion corrections to DFT, and the latter directly influences the computed equilibrium shapes (Wulff constructions). This study may serve as a starting point for future explorations of surfaces and nanocrystals of PbS, PbSe, and chemically related chalcogenide materials.
Co-reporter:Volker L. Deringer, Ralf P. Stoffel, Matthias Wuttig and Richard Dronskowski
Chemical Science 2015 vol. 6(Issue 9) pp:5255-5262
Publication Date(Web):29 Jun 2015
DOI:10.1039/C5SC00825E
Antimony selenide (antimonselite, Sb2Se3) is a versatile functional material with emerging applications in solar cells. It also provides an intriguing prototype to study different modes of bonding in solid chalcogenides, all within one crystal structure. In this study, we unravel the complex bonding nature of crystalline Sb2Se3 by using an orbital-based descriptor (the crystal orbital Hamilton population, COHP) and by analysing phonon properties and interatomic force constants. We find particularly interesting behaviour for the medium-range Sb⋯Se contacts, which still contribute significant stabilisation but are much softer than the “traditional” covalent bonds. These results have implications for the assembly of Sb2Se3 nanostructures, and bond-projected force constants appear as a useful microscopic descriptor for investigating a larger number of chalcogenide functional materials in the future.
Co-reporter:Volker L. Deringer, Wei Zhang, Pascal Rausch, Riccardo Mazzarello, Richard Dronskowski and Matthias Wuttig
Journal of Materials Chemistry A 2015 vol. 3(Issue 37) pp:9519-9523
Publication Date(Web):26 Aug 2015
DOI:10.1039/C5TC02314A
We identify a similar feature in the chemical-bonding nature of seemingly different phase-change materials (PCMs) for data storage. This affords new insight into the “next-generation” material In3SbTe2, establishes a hitherto missing link to the more ubiquitous Ge–Sb–Te alloys, and encourages the search for new PCMs beyond established electron-counting schemes.
Co-reporter:Janine George; Volker L. Deringer
Inorganic Chemistry 2015 Volume 54(Issue 3) pp:956-962
Publication Date(Web):November 3, 2014
DOI:10.1021/ic5023328
Two-dimensional (2D) and layered structures gained a lot of attention in the recent years (“post-graphene era”). The chalcogen cyanides S(CN)2 and Se(CN)2 offer themselves as interesting model systems to study layered inorganic crystal structures; both are built up from cyanide molecules connected by chalcogen bonds (ChBs). Here, we investigate ChBs and their cooperativity directly within the layers of the S(CN)2 and Se(CN)2 crystal structures and, furthermore, in putative O(CN)2 and Te(CN)2 crystal structures derived therefrom. Moreover, we determine the energetic contributions of ChBs within the layers to the overall stabilization energy. To compare these structures not only energetically but also geometrically, we derive a direction-dependent root mean square of the Cartesian displacement, a possible tool for further computational investigations of layered compounds. The molecular chains connected by ChBs are highly cooperative but do not influence each other when combined to layers: the ChBs are nearly orthogonal in terms of energy when connected to the same chalcogen acceptor atom. Layers built up from ChBs account for 41% to 79% of the overall interaction energy in the crystal. This provides new, fundamental insight into the meaning of ChBs, and therefore directed intermolecular interactions, for the stability of crystal structures.
Co-reporter:Tanja Scholz
Inorganic Chemistry 2015 Volume 54(Issue 17) pp:8800-8807
Publication Date(Web):August 19, 2015
DOI:10.1021/acs.inorgchem.5b01510
We report a synthetic and theoretical study of the solid solution SnxFe4–xN (0 ≤ x ≤ 0.9). A previously published ammonolytic synthesis was successfully modified to achieve the metastable nitrides in phase-pure quality out of many competing phases. As TG-DSC measurements show, the thermal stability of the nitrides increases with increasing tin content. The SnxFe4–xN series of compounds adopts an antiperovskite-like structure in space group Pm3̅m. Various experimental and theoretical methods provide evidence that the iron substitution by tin exclusively takes place at Wyckoff position 1a and leads to a Vegard-type behavior of the lattice parameter over the compositional range, with an expection for a small internal miscibility gap around Sn0.33Fe3.67N of unknown cause. For highly tin-substituted iron nitrides the composition was clarified by prompt gamma-ray activation analysis (PGAA) and determined as Sn0.78(3)Fe3.22(4)N0.95(3) evidencing a fully occupied nitrogen position. Magnetic measurements reveal a linear weakening of ferromagnetic interactions with increasing tin concentration.
Co-reporter:Janine George, Ai Wang, Volker L. Deringer, Ruimin Wang, Richard Dronskowski and Ulli Englert
CrystEngComm 2015 vol. 17(Issue 38) pp:7414-7422
Publication Date(Web):25 Aug 2015
DOI:10.1039/C5CE01219H
In chemical crystallography, the thermal motion of scattering centres is commonly described by anisotropic displacement parameters (ADPs). Very recently, it has been shown that ADPs are not only accessible by diffraction experiments but also via theory: this emerging approach seems promising but must be thoroughly tested. In this study, we have performed specifically tailored X-ray diffraction (XRD) experiments in fine steps between 100 and 300 K which allow detailed comparison to ab initio data from dispersion-corrected density functional theory (DFT) combined with periodic lattice-dynamics. The compound chosen for this study, crystalline pentachloropyridine (C5NCl5), is well suited for this purpose: it represents a molecular crystal without H atoms, thus posing no challenge to XRD; its solid-state structure is controlled by dispersion and halogen-bonding interactions; and the ADPs associated with the peripheral Cl atoms show strong temperature dependence. Quality criteria in direct and in reciprocal space prove that ADPs are predicted with high confidence for the temperature range between 100 and 200 K, and that several economic dispersion corrections to DFT can be reliably employed for this purpose. Within the limits we have explored here, the ab initio prediction of ADPs appears to be a facile and complementary tool, especially in those cases where diffraction data cannot provide a straightforward model for thermal motion.
Co-reporter:Marc Esser, Volker L. Deringer, Matthias Wuttig, Richard Dronskowski
Solid State Communications 2015 Volume 203() pp:31-34
Publication Date(Web):February 2015
DOI:10.1016/j.ssc.2014.11.008
Highlights•A new method for material characterization is presented, improving upon the concept of “material maps”.•Different functional materials are distinguished by falling into different areas of such maps.•The novel approach is able to discern polymorphs, adding structural information to such maps for the first time.•The maps are created fully quantum-mechanically from first-principles plane-wave/PAW DFT output.The creation of “maps” for solid-state materials has a long-standing history in condensed matter theory. Here, based on periodic density-functional theory (DFT) output, a heuristic numerical indicator is constructed to assess s–p orbital mixing in materials (or, depending on one׳s viewpoint, the tendency toward “sp3 hybridization”). Other than before, this now intrinsically includes structural information and the microscopic effects associated with it. The new method provides useful insights to understand physical relationships in composition space and promises to help to identify hitherto unknown material candidates.
Co-reporter:Dr. Xiaohui Liu;Janine George;M.Sc.Dipl.-Chem. Stefan Maintz;Dr. Richard Dronskowski
Angewandte Chemie 2015 Volume 127( Issue 6) pp:1977-1982
Publication Date(Web):
DOI:10.1002/ange.201410987
Abstract
Wir berichten über die Synthese einer unerwarteten Modifikation der hochgradig energiereichen Phase CuN3, die in der orthorhombischen Raumgruppe Cmcm mit a=3.3635(7), b=10.669(2), c=5.5547(11) Å und V=199.34(7) Å3 kristallisiert. Die schichtartige Struktur ähnelt Graphit mit einem Schichtabstand von 2.777(1) Å (=1/2 c). Sofern man N3− als strukturelle Einheit ansieht, liegen innerhalb einer Schicht annähernd hexagonale Zehnerringe mit heterographenartigem Motiv vor. Kupfer- und Stickstoffatome sind mit Cu-N=1×1.91 und 2×2.00 Å kovalent aneinander gebunden, und die N3−-Gruppe fällt mit N-N=1.14 und 1.20 Å linear aus. Berechnungen der Elektronenstrukturen und experimentelle Thermochemie ergeben, daß die als β-CuN3 bezeichnete neue Modifikation stabiler als die bekannte Phase α-CuN3 ist. Des weiteren erweist sich β-CuN3 anhand der berechneten phononischen Zustandsdichte als dynamisch und deshalb auch thermochemisch metastabil. β-CuN3 zeigt in den heterographenartigen Schichten eine negative thermische Ausdehnung.
Co-reporter:Dr. Volker L. Deringer;Dr. Richard Dronskowski
Angewandte Chemie 2015 Volume 127( Issue 51) pp:15550-15557
Publication Date(Web):
DOI:10.1002/ange.201506874
Abstract
Die Synthese und die Nutzbarmachung von Nanokristallen sind hochaktuelle Forschungsfelder, doch setzen sie ein gründliches Verständnis der beteiligten Kristallflächen voraus. In diesem Kurzaufsatz schlagen wir ebendiesen Bogen von Oberflächen zu freien und weiter zu naßchemisch synthetisierten Nanokristallen, und zwar exemplarisch für Bleiselenid (PbSe), Zinntellurid (SnTe) und ihre direkten chemischen Verwandten. Wir diskutieren experimentelle Einblicke und darüber hinaus die – zunehmend einflußreiche – quantenchemische Simulation von Oberflächen und Nanokristallen. Was kann die Theorie heute leisten, was möglicherweise morgen, was kann sie eben nicht? Die Beantwortung dieser Fragen und ihre geschickte Verknüpfung mit Experimenten könnten neue, atomistisch (und damit chemisch) geleitete Perspektiven in der Nanosynthese eröffnen.
Co-reporter:Dr. Xiaohui Liu;Janine George;M.Sc.Dipl.-Chem. Stefan Maintz;Dr. Richard Dronskowski
Angewandte Chemie International Edition 2015 Volume 54( Issue 6) pp:1954-1959
Publication Date(Web):
DOI:10.1002/anie.201410987
Abstract
An unexpected polymorph of the highly energetic phase CuN3 has been synthesized and crystallizes in the orthorhombic space group Cmcm with a=3.3635(7), b=10.669(2), c=5.5547(11) Å and V=199.34(7) Å3. The layered structure resembles graphite with an interlayer distance of 2.777(1) Å (=1/2 c). Within a single layer, considering N3− as one structural unit, there are 10-membered almost hexagonal rings with a heterographene-like motif. Copper and nitrogen atoms are covalently bonded with CuN bonds lengths of 1.91 and 2.00 Å, and the N3− group is linear but with NN 1.14 and 1.20 Å. Electronic-structure calculations and experimental thermochemistry show that the new polymorph termed β-CuN3 is more stable than the established α-CuN3 phase. Also, β-CuN3 is dynamically, and thus thermochemically, metastable according to the calculated phonon density of states. In addition, β-CuN3 exhibits negative thermal expansion within the graphene-like layer.
Co-reporter:Dr. Volker L. Deringer;Dr. Richard Dronskowski
Angewandte Chemie International Edition 2015 Volume 54( Issue 51) pp:15334-15340
Publication Date(Web):
DOI:10.1002/anie.201506874
Abstract
Synthesis and utilization of nanocrystals are highly active fields of current research, but they require a thorough understanding of the underlying crystal surfaces. In this Minireview, we span the arc from surfaces to free nanocrystals, and onward to their chemical synthesis, using as examples lead selenide (PbSe), tin telluride (SnTe), and their direct chemical relatives. Besides experimental insights, we highlight the increasingly influential role played by quantum-chemical simulations of surfaces and nanocrystals. What can theory do today, or possibly tomorrow; where are its limits? Answering these questions, and skillfully linking them to experiments, could open up new atomistically (that is, chemically) guided perspectives for nanosynthesis.
Co-reporter:Janine George;Christoph Reimann;Dr. Volker L. Deringer; Dr. Thomas Bredow; Dr. Richard Dronskowski
ChemPhysChem 2015 Volume 16( Issue 4) pp:728-732
Publication Date(Web):
DOI:10.1002/cphc.201402890
Abstract
We report on an erroneous ground state within common density functional theory (DFT) methods for the solid elements bromine and iodine. Phonon computations at the GGA level for both molecular crystals yield imaginary vibrational modes, erroneously indicating dynamic instability—that fact alone could easily pass as a computational artefact, but these imaginary modes lead to energetically more favorable and dynamically stable structures, made up of infinite monoatomic chains. In contrast, meta-GGA and hybrid functionals yield the correct energetic order for bromine, while for iodine, most global hybrids do not improve the GGA result significantly. The qualitatively correct answer, in both cases, is given by the long-range corrected hybrid LC-ωPBE, the Minnesota functionals M06L and M06, and by periodic Hartree–Fock and MP2 theory. This poor performance of economic DFT functionals should be kept in mind, for example, during global structure optimizations of systems with significant contributions from halogen bonds.
Co-reporter:Janine George;Christoph Reimann;Dr. Volker L. Deringer; Dr. Thomas Bredow; Dr. Richard Dronskowski
ChemPhysChem 2015 Volume 16( Issue 4) pp:
Publication Date(Web):
DOI:10.1002/cphc.201590021
Co-reporter:M.Sc. Ronja Missong;M.Sc. Janine George;Dr. Andreas Houben;Dr. Markus Hoelzel;Dr. Richard Dronskowski
Angewandte Chemie International Edition 2015 Volume 54( Issue 41) pp:12171-12175
Publication Date(Web):
DOI:10.1002/anie.201507113
Abstract
Strontium guanidinate, SrC(NH)3, the first compound with a doubly deprotonated guanidine unit, was synthesized from strontium and guanidine in liquid ammonia and characterized by X-ray and neutron diffraction, IR spectroscopy, and density-functional theory including harmonic phonon calculations. The compound crystallizes in the hexagonal space group P63/m, constitutes the nitrogen analogue of strontium carbonate, SrCO3, and its structure follows a layered motif between Sr2+ ions and complex anions of the type C(NH)32−; the anions adopt the peculiar trinacria shape. A comparison of theoretical phonons with experimental IR bands as well as quantum-chemical bonding analyses yield a first insight into bonding and packing of the formerly unknown anion in the crystal.
Co-reporter:M.Sc. Ronja Missong;M.Sc. Janine George;Dr. Andreas Houben;Dr. Markus Hoelzel;Dr. Richard Dronskowski
Angewandte Chemie 2015 Volume 127( Issue 41) pp:12339-12343
Publication Date(Web):
DOI:10.1002/ange.201507113
Abstract
Strontiumguanidinat, SrC(NH)3, die erste Verbindung mit zweifach deprotonierter Guanidineinheit, wurde aus Strontium und Guanidin in flüssigem Ammoniak synthetisiert und mittels Röntgen- und Neutronenpulverdiffraktion, IR-Spektroskopie und Dichtefunktionaltheorie inklusive harmonischer Phononenrechnungen charakterisiert. Die in der hexagonalen Raumgruppe P63 /m kristallisierende Verbindung ist das Stickstoffanalogon des Strontiumcarbonats, SrCO3, und dem Motiv nach schichtartig aus Sr2+-Ionen und komplexen Anionen des Typs C(NH)32− aufgebaut; letztere nehmen die eigentümliche Trinacriagestalt an. Der Vergleich theoretischer Phononenrechnungen mit experimentellen IR-Banden sowie quantenchemische Bindungsanalysen erlauben einen ersten Einblick in die Bindungs- und Packungsverhältnisse des bisher unbekannten Anions im Kristall.
Co-reporter:Volker L. Deringer and Richard Dronskowski
Chemical Science 2014 vol. 5(Issue 3) pp:894-903
Publication Date(Web):15 Nov 2013
DOI:10.1039/C3SC52743C
Germanium dioxide (GeO2) finds increasing application on the nanoscale, which calls for a detailed understanding of its crystal surfaces. In particular, the metastable α-quartz-type polymorph of GeO2 exhibits many desirable properties but also a non-trivial structural chemistry. Here, we contribute a surface study of quartz-type GeO2 in which we combine periodic density-functional theory (DFT) with classical chemical reasoning. We report on the most relevant surfaces, both freshly cleaved and structurally optimised. Stability trends of the latter are discussed in terms of the central structural unit—the [GeO4] tetrahedra—and how they are linked at the surface, in seamless extension of Pauling's third rule which had originally been conceived for bulk crystal structures. A more detailed, energy-resolved view is afforded by computing crystal orbital overlap populations (COOP) with a novel projection scheme; this way, a “bond strength” is directly gauged from plane-wave DFT output, and it allows the different surface stabilities to be rationalised in terms of “strengthened” and “weakened” bonds. These results and ways of thinking may be relevant for future studies on nanocrystalline GeO2 and, in a broader context, also for silica (SiO2) and other surfaces.
Co-reporter:Volker L. Deringer, Ulli Englert and Richard Dronskowski
Chemical Communications 2014 vol. 50(Issue 78) pp:11547-11549
Publication Date(Web):08 Aug 2014
DOI:10.1039/C4CC04716H
The covalent nature of short hydrogen bonds has been under debate for long. Here we show that the crystal orbital Hamilton population (COHP) bonding indicator gives new, complementary evidence of covalent hydrogen⋯acceptor interactions in the molecular solid state.
Co-reporter:Marck Lumeij, Michael Gilleßen, Henny Bouwmeester, Torsten Markus, Juri Barthel, Stefan Roitsch, Joachim Mayer and Richard Dronskowski
Physical Chemistry Chemical Physics 2014 vol. 16(Issue 4) pp:1333-1338
Publication Date(Web):20 Nov 2013
DOI:10.1039/C3CP53958J
We present a theoretical and experimental study on the influence of the Ba/Sr and Co/Fe ratios as well as the oxygen-non-stoichiometry on the stability of Ba0.5Sr0.5Co0.8Fe0.2O3−δ (BSCF). Thin-layer depositions are analysed by looking at TEM images and EDX spectra. Bond-analytical calculations are performed to explain the stability difference between hexagonal and cubic BSCF. Finally, annealing experiments analysed using XRD give an insight into the differences of phase-fraction growth with respect to the Ba/Sr ratio.
Co-reporter:Volker L. Deringer, Ralf P. Stoffel, and Richard Dronskowski
Crystal Growth & Design 2014 Volume 14(Issue 2) pp:871-878
Publication Date(Web):January 13, 2014
DOI:10.1021/cg401822g
The ability to predict temperature-dependent properties of solids from ab initio theories alone has marked a fundamental step forward in realistic materials modeling. Here, we use density functional theory (DFT)-based thermochemistry to investigate crystalline tellurium dioxide (TeO2). While the DFT description of its polymorphs is challenging, we demonstrate that the inclusion of suitable dispersion corrections makes a thermochemical analysis and a correct stability ranking possible. Paratellurite (α-TeO2), a technologically most important compound used in optical devices, is lowest in Gibbs free enthalpy in agreement with previous thermochemical experiments. While α-TeO2 exhibits a three-dimensional bonding network, the well-known mineral tellurite (β-TeO2) contains two-dimensional sheets; nonetheless, we demonstrate that both polymorphs are extremely similar in thermodynamic state functions and chemical bonding. The latter is analyzed with the crystal orbital Hamilton population (COHP) method and offered as a possible explanation for the small stability difference (<1 kJ mol–1). Finally, a recently synthesized polymorph (γ-TeO2) is found to be higher in energy as expected (+6 kJ mol–1 at 300 K) but nonetheless dynamically stable. These results shed new light on the TeO2 polymorphs but also provide more general insight into the capabilities of theoretical materials modeling at finite temperatures.
Co-reporter:Volker L. Deringer, Ralf P. Stoffel, Atsushi Togo, Bernhard Eck, Martin Meven and Richard Dronskowski
CrystEngComm 2014 vol. 16(Issue 47) pp:10907-10915
Publication Date(Web):21 Oct 2014
DOI:10.1039/C4CE01637H
The thermal motion of atoms and functional groups is a key characteristic of any molecular crystal, and such motion derived from scattering experiments is conveniently visualised by means of thermal ellipsoids (the famous “ORTEP” drawings). Unfortunately, it is often impossible to obtain the underlying anisotropic displacement parameters (ADPs) for hydrogen atoms, due to their low X-ray scattering power, and sometimes no ADPs can be refined at all even for heavier atoms. In these cases, it would seem advantageous to estimate ADPs by first-principles techniques, and indeed such ab initio ORTEP plots have become available very recently. Here, we test this young method for a representative set of hydrogen-bonded molecular crystals: first, we study urea (CON2H4) as a well-known benchmark, then, its all-nitrogen analogue guanidine (CN3H5); finally, we move on to rubidium guanidinate (RbCN3H4) as a specimen with pronounced ionic interactions. For all three systems, ADPs have been obtained from density-functional theory (DFT) based phonon computations using the PHONOPY software. The results are compared with neutron-diffraction data as the experimental “benchmark” in this regard, and a critical discussion of experimental aspects is given. We observe excellent agreement between experiment and theory for the hydrogen-bonded systems urea and guanidine at low temperature, whereas high-temperature data for guanidine deviate visibly, and the more salt-like RbCN3H4 may suffer from a less-than-ideal description even at 12 K. Both are discussed in depth as there are possible solutions and directions for further research. Generally, the present results shine a favourable light on a future, more routine application of combined experimental/theoretical approaches in chemical crystallography.
Co-reporter:Volker L. Deringer, Fangfang Pan, Janine George, Paul Müller, Richard Dronskowski and Ulli Englert
CrystEngComm 2014 vol. 16(Issue 2) pp:135-138
Publication Date(Web):11 Nov 2013
DOI:10.1039/C3CE41779D
Mutually perpendicular hydrogen bonds, halogen bonds and stacking interactions are investigated in the seemingly simple crystal structure of bromomalonic aldehyde (C3H3O2Br). This combined experimental and theoretical study sheds new light on the role of electrostatic interactions in molecular crystalline networks and poses a caveat to look beyond what may be termed “chemical intuition”.
Co-reporter:Tobias A. Timmerscheidt, Jörg von Appen, Richard Dronskowski
Computational Materials Science 2014 Volume 91() pp:235-239
Publication Date(Web):August 2014
DOI:10.1016/j.commatsci.2014.04.054
•Carbon diffusion in face-centered cubic iron is studied by molecular-dynamics simulations.•The Modified Embedded Atom Method (MEAM) potential for the Fe–C system by Lee is used.•Diffusion is studied for carbon concentrations of 0.6, 1.1 and 1.4 wt%.•Based on an Arrhenius approach the results are compared to empirical models.Molecular-dynamics calculations targeted at the diffusion of carbon in γ-iron were performed using the Modified Embedded Atom Method (MEAM) interatomic potential by Lee. The diffusion coefficients were calculated at different temperatures and carbon concentrations. A temperature-dependence of the diffusion coefficient according to the Arrhenius law was assumed. By doing so, activation energies as well as pre-exponential factors for different carbon concentrations were derived from the diffusion coefficients and compared to experimental values. Good agreement was reached for the activation energies while the calculated pre-exponential factors differ from experimental values.Graphical abstract
Co-reporter:Dipl.-Chem. Volker L. Deringer;Dr. Wei Zhang;Dr. Marck Lumeij;Dipl.-Chem. Stefan Maintz;Dr. Matthias Wuttig;Dr. Riccardo Mazzarello;Dr. Richard Dronskowski
Angewandte Chemie International Edition 2014 Volume 53( Issue 40) pp:10817-10820
Publication Date(Web):
DOI:10.1002/anie.201404223
Abstract
Despite its simple chemical constitution and unparalleled technological importance, the phase-change material germanium telluride (GeTe) still poses fundamental questions. In particular, the bonding mechanisms in amorphous GeTe have remained elusive to date, owing to the lack of suitable bond-analysis tools. Herein, we introduce a bonding indicator for amorphous structures, dubbed “bond-weighted distribution function” (BWDF), and we apply this method to amorphous GeTe. The results underline a peculiar role of homopolar GeGe bonds, which locally stabilize tetrahedral fragments but not the global network. This atom-resolved (i.e., chemical) perspective has implications for the stability of amorphous “zero bits” and thus for the technologically relevant resistance-drift phenomenon.
Co-reporter:Dipl.-Chem. Volker L. Deringer;Dr. Wei Zhang;Dr. Marck Lumeij;Dipl.-Chem. Stefan Maintz;Dr. Matthias Wuttig;Dr. Riccardo Mazzarello;Dr. Richard Dronskowski
Angewandte Chemie International Edition 2014 Volume 53( Issue 40) pp:
Publication Date(Web):
DOI:10.1002/anie.201406993
Co-reporter:Janine George, Volker L. Deringer, and Richard Dronskowski
The Journal of Physical Chemistry A 2014 Volume 118(Issue 17) pp:3193-3200
Publication Date(Web):April 8, 2014
DOI:10.1021/jp5015302
Halogen bonds (XBs) are intriguing noncovalent interactions that are frequently being exploited for crystal engineering. Recently, similar bonding mechanisms have been proposed for adjacent main-group elements, and noncovalent “chalcogen bonds” and “pnictogen bonds” have been identified in crystal structures. A fundamental question, largely unresolved thus far, is how XBs and related contacts interact with each other in crystals; similar to hydrogen bonding, one might expect “cooperativity” (bonds amplifying each other), but evidence has been sparse. Here, we explore the crucial step from gas-phase oligomers to truly infinite chains by means of quantum chemical computations. A periodic density functional theory (DFT) framework allows us to address polymeric chains of molecules avoiding the dreaded “cluster effects” as well as the arbitrariness of defining a “large enough” cluster. We focus on three types of molecular chains that we cut from crystal structures; furthermore, we explore reasonable substitutional variants in silico. We find evidence of cooperativity in chains of halogen cyanides and also in similar chalcogen- and pnictogen-bonded systems; the bonds, in the most extreme cases, are amplified through cooperative effects by 79% (I···N), 90% (Te···N), and 103% (Sb···N). Two experimentally known organic crystals, albeit with similar atomic connectivity and XB characteristics, show signs of cooperativity in one case but not in another. Finally, no cooperativity is observed in alternating halogen/acetone and halogen/1,4-dioxane chains; in fact, these XBs weaken each other by up to 26% compared to the respective gas-phase dimers.
Co-reporter:Volker L. Deringer, Marck Lumeij, Ralf P. Stoffel, and Richard Dronskowski
Chemistry of Materials 2013 Volume 25(Issue 11) pp:2220
Publication Date(Web):May 9, 2013
DOI:10.1021/cm400316j
Germanium telluride (GeTe) is an iconic functional material, both in itself and as the parent compound for a range of ternary phase-change data-storage alloys. Long taken to be a “simple” AB compound, crystalline GeTe is today known to contain a large number of germanium vacancies which directly affect the material’s macroscopic properties. Here, we use atomistic simulations to elucidate local mechanisms behind the motion of Ge atoms (and thus, vacancy diffusion) in crystalline GeTe. Transition pathways are computed using the nudged elastic band (NEB) approach at the gradient-corrected level of density-functional theory (GGA-DFT), both for the idealized rhombohedral (R3m) crystal and a number of defective configurations. Besides obvious structural arguments (i.e., beyond a simple rigid-sphere model), the diffusion barriers show a delicate dependence on the material’s electronic structure. The latter is controlled by vacancy formation, Sb adatoms, and charge injection, all of which is discussed in a unified framework.Keywords: density-functional theory (DFT); GST alloys; nudged elastic band (NEB); phase-change materials;
Co-reporter:Peter Klaus Sawinski, Volker L. Deringer and Richard Dronskowski
Dalton Transactions 2013 vol. 42(Issue 42) pp:15080-15087
Publication Date(Web):13 Aug 2013
DOI:10.1039/C3DT51820E
A family of alkali-metal guanidinates (M = Na–Cs) has been reported recently, representing all-nitrogen analogues of alkali bicarbonates and containing the elusive guanidinate monoanion CN3H4−. Here, we describe the synthesis and characterisation of LiCN3H4 as the last representative of alkali guanidinates. Single crystals of LiCN3H4 were obtained using a solvothermal route in liquid ammonia, and the crystal structure was determined at 100 K using single-crystal X-ray diffraction (monoclinic, P21/c, Z = 4, a = 7.251(2) Å, b = 4.532(2) Å, c = 9.051(2) Å, β = 103.315(3)°). The structure of LiCN3H4 is reminiscent of the previously reported NaCN3H4 type in that both guanidinates contain tetrahedrally nitrogen-coordinated alkali-metal cations. The linking of these tetrahedra differs, however: they share corners in NaCN3H4 to form one-dimensionally infinite chains running through the crystal, whereas isolated, edge-sharing tetrahedra (Li2N6 motif) are found in the LiCN3H4 structure described here. Periodic density-functional theory (DFT) computations at the dispersion-corrected PBE + D2 level not only correctly reproduce the structural preferences, but also indicate that a NaCN3H4 polymorph should be viable adopting the lithium guanidinate type but much less easily so vice versa.
Co-reporter:Peter Klaus Sawinski, Martin Meven, Ulli Englert, and Richard Dronskowski
Crystal Growth & Design 2013 Volume 13(Issue 4) pp:1730-1735
Publication Date(Web):February 14, 2013
DOI:10.1021/cg400054k
Pure guanidine crystallizes in the orthorhombic space group Pbca (no. 61) and a = 8.5022(2) Å, b = 9.0863(2) Å, c = 15.6786(4) Å at 100 K, Z = 16, with two Y-shaped molecules in the asymmetric unit. The compound features a three-dimensional network of classical N–H···N hydrogen bonds. Here, we present the results of a single-crystal neutron diffraction study, performed at two different temperatures (100 and 273 K). The data quality obtained at the HEiDi instrument (FRM II, Munich) allowed to derive accurate positional and anisotropic displacement parameters (ADP) for all the atoms, including H. The experimental hydrogen positions confirm a model derived from theory. On the basis of the displacement parameters, a TLS analysis of thermal motion proves that the guanidine molecules behave in good approximation as rigid bodies and essentially undergo libration. The unusual temperature behavior of one C–N bond found in a preceding single-crystal X-ray study is an artifact going back to this rigid-body movement. The existence of various hydrogen bonds also manifests from a well-resolved IR spectrum, which was analyzed in terms of individual vibrations on the basis of quasi-harmonic ab initio phonon calculations.
Co-reporter:Sarah Lintzen, Jörg von Appen, Bengt Hallstedt, Richard Dronskowski
Journal of Alloys and Compounds 2013 Volume 577() pp:370-375
Publication Date(Web):15 November 2013
DOI:10.1016/j.jallcom.2013.06.006
Highlights•A quantum-theoretical phase diagram including three structure types has been generated.•The magnetic ground states of various stoichiometries have been tested.•The course of the volumes as a function of the composition has been analyzed.•A comparison between DFT and CALPHAD has been carried out.Density-functional theory-based total-energy calculations have been performed in order to ascertain the heats of formation of the binary phases Fe1–xMnx in the bcc, fcc, and hcp structure types. Each structure type and composition range of the solid solutions have been critically assessed with respect to their magnetic ground state and the course of the molar volume as a function of the stoichiometry. All theoretical energy data have been combined into an enthalpy–composition phase diagram, to be critically compared with an analogous plot derived from the CALPHAD method. The two radically different methods result in very similar diagrams. Noticeable deviations do occur as well, particularly for highly unstable regions where the real magnetic ground state is a matter of debate.Graphical abstract
Co-reporter:C. Wessel, R. Dronskowski
Journal of Solid State Chemistry 2013 Volume 199() pp:149-153
Publication Date(Web):March 2013
DOI:10.1016/j.jssc.2012.12.019
Chromium sesquioxide has been investigated by the density-functional theory (GGA/PBE/PAW). This study focuses on the experimental high-pressure polymorph, which until now could not be characterized satisfactorily. The eligible structures for high-pressure Cr2O3, [Rh2O3(II)] and the orthorhombic perovskite [GdFeO3], are compared to the ambient-pressure corundum polymorph. The analysis yields the [Rh2O3(II)] structure as the most likely high-pressure phase while phonon calculations verify its dynamic stability. Additionally, the cubic [c-type Ln2O3], dubbed bixbyite, is identified as a possible ambient-pressure polymorph.Graphical abstractA theoretical high-pressure study on chromium sesquioxide indicates that Cr2O3 is likely to adopt the Rh2O3(II) structure type at high pressure, and the latter structure appears as a minimum on the potential energy surface.Highlights► Ab initio calculations predict the [Rh2O3(II)] type as a possible Cr2O3 high-pressure polymorph. ► Phonon calculations indicate that Cr2O3 adopting the [Rh2O3(II)] type is dynamically stable. ► The calculation of the effective coordination number according to Brunner and Schwarzenbach yields a preferred coordination number larger than 5.5 for high-pressure Cr2O3. ► The bixbyite type is identified as an energetically promising candidate for a new Cr2O3 polymorph at ambient pressure.
Co-reporter:Volker L. Deringer; Dr. Richard Dronskowski
ChemPhysChem 2013 Volume 14( Issue 13) pp:3108-3111
Publication Date(Web):
DOI:10.1002/cphc.201300265
Co-reporter:Volker L. Deringer and Richard Dronskowski
The Journal of Physical Chemistry C 2013 Volume 117(Issue 29) pp:15075-15089
Publication Date(Web):July 8, 2013
DOI:10.1021/jp401400k
Ternary alloys of germanium, antimony, and tellurium (GexSbyTez) are not only prominent phase-change data-storage materials with intriguing properties; they also form precisely ordered crystal structures if one allows them to do so. Here, we study atomically pristine surfaces of representative GexSbyTez alloys using density functional theory (DFT) and suitable plane-wave-based slab models. In particular, we look at the hexagonal (0001) surfaces of Ge1Sb2Te4, Ge2Sb2Te5, and Ge1Sb4Te7 but with the intent to draw some general conclusions beyond those explicit compositions. The conditions for thermodynamic stability of the ternary surfaces (i.e., the space of permissible chemical potentials) are delineated, and a simple general expression to estimate the latter is given. Because of weak van der Waals-type interactions between Te atomic layers, we assess how surface energy computations are affected by the choice of exchange–correlation functional and also by the use of dispersion corrections (here, Grimme’s DFT+D2 scheme). LDA and PBE+D2 predicted surface energies are similar to a degree that is rather unexpected; generally, Te terminations are lowest in surface energy regardless of the compound and methodology. Implications for further surface studies of “less ideal’’ GexSbyTez surfaces are discussed.
Co-reporter:Volker L. Deringer and Richard Dronskowski
The Journal of Physical Chemistry C 2013 Volume 117(Issue 46) pp:24455-24461
Publication Date(Web):October 28, 2013
DOI:10.1021/jp408699a
Lead telluride (PbTe) is a well-known functional material with current applications in fields such as nanostructured thermoelectrics. Here, we report comprehensive first-principles simulations for important crystal surfaces of rocksalt-type PbTe. Surface energies have been computed for pristine (001), (011), and (111) surfaces, and a number of relevant (111) surface reconstructions have been explored. Density-functional theory (DFT) was applied at various levels of sophistication and cost, namely, the local density approximation (LDA), the generalized gradient approximation (GGA), and the HSE06 hybrid functional. Overall, PbTe(001) is predicted to be noticeably stable, essentially corroborating previous computational studies that had been performed for this particular termination; here, however, we expand upon these findings and use the computed set of surface-energy data to predict the equilibrium crystallite shape (as reflected by the Wulff construction); the latter is strongly dominated by {001} facets throughout the entire chemical-potential space. For the PbTe(111) surface, atomic reconstructions are investigated, and they emerge as energetically more favorable than either of the two pristine terminations: this in contrast to the lighter homologues GeTe and SnTe. In particular, for PbTe, 2 × 1 and 2 × 2 reconstructions are predicted and assessed in comprehensive surface phase diagrams. The computational results compare favorably to previous experiments including evidence for a 2 × 2 reconstruction of the (111) surface; overall, they may contribute a useful piece of understanding of these important crystal surfaces.
Co-reporter:Michael Gilleßen, Marck Lumeij, Janine George, Ralf Stoffel, Teruki Motohashi, Shinichi Kikkawa, and Richard Dronskowski
Chemistry of Materials 2012 Volume 24(Issue 10) pp:1910
Publication Date(Web):May 3, 2012
DOI:10.1021/cm300655y
The experimentally known perovskite-like materials BaYMn2O5+δ (δ = 0, 0.5, 1) are characterized by a remarkably reversible oxygen-storage capacity at a moderate 500 °C. We try to elucidate the local structures of the vacancy arrangements in these compounds taking place after an oxygen release. This is done for the three compounds with the help of both ab initio total-energy calculations of density-functional quality and using classical structure rationale. Our results are compared with experimental structure findings. We further calculate oxygen-vacancy formation energies and predict the pathways of the oxygen atoms through the crystal by using NEB (nudged elastic band) calculations. Structure diagrams of the most likely energy pathways for oxygen migration are presented. Finally, thermodynamic considerations of the oxygen intake are carried out based on quasiharmonic phonon calculations and compared with experimental data. The theoretical molar reaction enthalpy for oxidizing BaYMn2O5 to BaYMn2O6 matches the experimental value.Keywords: ab initio thermochemistry; BaYMn2O6; DFT; nudged elastic band; oxygen hopping pathways; oxygen-storage material;
Co-reporter:Peter Klaus Sawinski and Richard Dronskowski
Inorganic Chemistry 2012 Volume 51(Issue 13) pp:7425-7430
Publication Date(Web):June 19, 2012
DOI:10.1021/ic301005x
Phase-pure NaCN3H4 and KCN3H4 were synthesized from molecular guanidine and elemental metal in liquid ammonia at room temperature and elevated pressure close to 10 atm. The crystal structures were determined at 100 K using single-crystal X-ray diffraction. Both compounds crystallize in the monoclinic system (P21/c, No. 14) but are far from being isotypical. NaCN3H4 (a = 7.9496(12) Å, b = 5.0328(8) Å, c = 9.3591(15) Å, β = 110.797(3)°, Z = 4) contains a tetrahedrally N-coordinated sodium cation while KCN3H4 (a = 7.1200(9) Å, b = 6.9385(9) Å, c = 30.404(4) Å, β = 94.626(2)°, Z = 16) features a very large c axis and a rather complicated packing of irregularly N-coordinated potassium cations. In the crystal structures, the guanidinate anions resemble the motif known from RbCN3H4, that is, with one elongated C–(amino)N single bond and two shorter C–(imino)N bonds (bond order = 1.5) although the orientation of one N–H bond differs in the guanidinate anion of NaCN3H4. Both crystal structures and infrared spectroscopy evidence the presence of hydrogen-bridging bonds, and the vibrational properties were analyzed by ab initio phonon calculations.
Co-reporter:Volker L. Deringer, Veronika Hoepfner, and Richard Dronskowski
Crystal Growth & Design 2012 Volume 12(Issue 2) pp:1014-1021
Publication Date(Web):December 16, 2011
DOI:10.1021/cg201505n
Organic molecules crystallize in manifold structures. The last few decades have seen the rise of high-resolution X-ray diffraction techniques that make the structures of even the most complex crystals easily accessible. Still, an intrinsic challenge lies in assigning hydrogen atoms’ positions from X-ray experiments alone. Quantum chemistry plays a fruitful, complementary role here, and so ab initio optimization techniques for organic crystals are on the rise as well. In this context, we review and evaluate a popular ab initio strategy based on plane-wave density-functional computations, namely, selectively relaxing H positions in an otherwise fixed cell. Our data show that such-optimized C–H, N–H, O–H, and B–H bond lengths coincide well with results from neutron diffraction—the experimental technique that sets the “gold standard” for H positions in molecular crystals but which is far less easily available. We have thus justified the use of a quantum-chemical aide with a broad variety of possible applications.
Co-reporter:Veronika Hoepfner, Volker L. Deringer, and Richard Dronskowski
The Journal of Physical Chemistry A 2012 Volume 116(Issue 18) pp:4551-4559
Publication Date(Web):March 15, 2012
DOI:10.1021/jp2106132
Hydrogen bonding is among the most important interactions in molecular crystals, and examples are abundant. As a consequence of such interactions, many molecules crystallize in complex but intriguing structures, in contrast to the relatively simple packing principles of metallic or ionic solids. In this work, we present a computational approach based on plane-wave density-functional theory (DFT) and supercell techniques, aiming to understand and quantify hydrogen-bonded networks in the solid state and in two-, one-, and zero-dimensional fragments derived from the molecular crystal. With such methodology at hand, we investigate guanidine, a fitting example of a molecular crystal and an important compound for inorganic and organic chemistry alike. On the basis of our computations, we discuss the initially proposed layered structure of guanidine and identify both stabilizing and destabilizing cooperative interactions in the three crystalline dimensions.
Co-reporter:Marck Lumeij, Julius Koettgen, Michael Gilleßen, Takanori Itoh, Richard Dronskowski
Solid State Ionics 2012 Volumes 222–223() pp:53-58
Publication Date(Web):20 August 2012
DOI:10.1016/j.ssi.2012.07.004
Co-reporter:Andrei L. Tchougréeff, Xiaohui Liu, Paul Müller, Wouter van Beek, Uwe Ruschewitz, and Richard Dronskowski
The Journal of Physical Chemistry Letters 2012 Volume 3(Issue 22) pp:3360-3366
Publication Date(Web):October 31, 2012
DOI:10.1021/jz301722b
We present the results of a high-resolution low-temperature synchrotron structural study of the nitrogen-containing analogue of cupric oxide, copper carbodiimide (CuNCN). The material clearly manifests a surprising nonmonotonous behavior of the a lattice parameter as a function of the temperature: its decrease with decreasing temperature turns to a distinguishable increase below 100 K. The onset temperature of this anomaly nearly coincides with that of the onset of the activation temperature dependence, as seen from the magnetic susceptibility, which has been tentatively attributed to the formation of a gapped 2D spin-liquid (resonating-valence-bond) state. In accord with this assumption, we develop a theory relating the temperature behavior of the spin susceptibility to that of the lattice parameter, which semiquantitatively agrees with the observed behavior.Keywords: carbodiimide; copper; crystal structure; resonating-valence bond theory; spin liquid; synchrotron study;
Co-reporter:Volker L. Deringer, Marck Lumeij, and Richard Dronskowski
The Journal of Physical Chemistry C 2012 Volume 116(Issue 29) pp:15801-15811
Publication Date(Web):June 22, 2012
DOI:10.1021/jp304455z
Density-functional theory (DFT) computations are reported for the (111) crystal surfaces of the phase-change material germanium telluride in its stable rhombohedral modification (dubbed α-GeTe). Atomic structures and surface energies are evaluated using a custom-tailored slab model and periodic plane-wave basis sets in the PBE-GGA approximation. Independent of the chemical surrounding, a pristine Te-covered (111) surface is energetically favorable among competing models, whereas a purely Ge-terminated surface is about 60 meV Å–2 higher in energy and predicted to undergo a structural reconstruction. Several atomic motifs for such reconstructions lie closely together energetically and are expected to coexist at finite temperatures. Formation of Ge vacancies in the subsurface layers is investigated in detail. Scanning tunneling microscopy (STM) images are simulated from the DFT wave functions for comparison with experiments to be performed in the foreseeable future.
Co-reporter:Michael Krings, Giuseppe Montana, Richard Dronskowski, and Claudia Wickleder
Chemistry of Materials 2011 Volume 23(Issue 7) pp:1694
Publication Date(Web):March 10, 2011
DOI:10.1021/cm102262u
Yellow Eu2+-doped strontium carbodiimide, SrNCN:Eu2+, adopting the α-SrNCN structure type was obtained by the reaction of SrI2, EuI2, CsN3 and CsCN in arc-welded Ta ampules. The product was characterized by high-resolution X-ray powder diffraction and infrared spectroscopy. Even at room temperature α-SrNCN:Eu2+ shows a strong orange emission peaking at 603 nm which is excitable by energies below 25 000 cm−1. The little change of the optical properties with increasing temperature is rather unexpected and leads to the assumption that this material is a promising candidate for future phosphor converted LEDs as well as a key compound for the understanding of the influence of the host lattice on the luminescence properties of Eu2+-doped materials.Keywords: Eu2+; LED; luminescence; phosphor; strontium carbodiimide;
Co-reporter:Veronika Hoepfner
Inorganic Chemistry 2011 Volume 50(Issue 8) pp:3799-3803
Publication Date(Web):March 11, 2011
DOI:10.1021/ic2002089
Rubidium guanidinate, RbCN3H4, was synthesized from guanidine and rubidium hydride, and the crystal structure was determined from powder X-ray diffraction (PXRD) data. RbCN3H4 crystallizes in the orthorhombic space group Pnma (No. 62) with four formula units per cell. The guanidinate anions are arranged in double chains running along the b axis, stacked almost perpendicularly to each other to form a three-dimensional network. The rubidium cations, coordinated by 11 N atoms, occupy the vacancies of the network in a zigzag motif along the b axis. Because the PXRD structure of the CN3 core clearly indicates the N-atom functionalities and the location of the H-atom positions, the latter spatial parameters were determined from Perdew−Burke−Ernzerhof generalized gradient approximation (GGA-PBE) density functional theory calculations. The corresponding ν(NH) stretching modes can be observed in the IR spectrum, and the volume chemistry of RbCN3H4 mirrors the efficient packing of the saltlike phase.
Co-reporter:Jens Burghaus, Moulay T. Sougrati, Anne Möchel, Andreas Houben, Raphaël P. Hermann, Richard Dronskowski
Journal of Solid State Chemistry 2011 Volume 184(Issue 9) pp:2315-2321
Publication Date(Web):September 2011
DOI:10.1016/j.jssc.2011.06.031
Prior investigations of the ternary nitride series Ga1–xFe3+xN (0≤x≤1) have indicated a transition from ferromagnetic γ′-Fe4N to antiferromagnetic “GaFe3N”. The ternary nitride “GaFe3N” has been magnetically and spectroscopically reinvestigated in order to explore the weakening of the ferromagnetic interactions through the gradual incorporation of gallium into γ′-Fe4N. A hysteretic loop at RT reveals the presence of a minority phase of only 0.1–0.2 at%, in accord with the sound two-step synthesis. The composition of the gallium-richest phase “GaFe3N” was clarified by Prompt Gamma-ray Activation Analysis and leads to the berthollide formula Ga0.91(1)Fe3.09(10)N1.05(7). Magnetic measurements indicate a transition around 8 K, further supported by Mössbauer spectral data. The weakening of the ferromagnetic coupling through an increasing gallium concentration is explained by a simple Stoner argument. In Ga0.9Fe3.1N, the presence of iron on the gallium site affects the magnetism by the formation of 13-atom iron clusters.Graphical AbstractThe crystal structure of GaFe3N with green nitrogen atoms in the very center, red iron atoms at the face centers, and gray gallium atoms at the corner positions.Highlights► Almost phase-pure synthesis of Ga0.9Fe3.1N. ► Prompt gamma-ray activation analysis yields precise composition. ► Magnetic ordering of the facial Fe atoms at the lowest temperatures. ► Mößbauer spectroscopy suggests percolation or RKKY-type interaction. ► Fe13 clusters due to berthollide character.
Co-reporter:Dr. Michael Wessel ;Dr. Richard Dronskowski
Chemistry - A European Journal 2011 Volume 17( Issue 9) pp:2598-2603
Publication Date(Web):
DOI:10.1002/chem.201003143
Abstract
Among numerous different AB2 structures with the hypothetical composition FeN2, the structures lying lowest in energy have been determined by a series of density-functional electronic-structure calculations. The most likely FeN2 phase crystallizing in the space group Rm must be considered an iron pernitride incorporating binuclear NN units (d=1.275 Å) with an anionic charge of 2−. This high-pressure magnetic phase with a bulk modulus of about 192 GPa and an iron saturation moment of approximately 1.68 μB should already form at a pressure of 17 GPa at an assumed reaction temperature of 1000 K. Besides bonding FeN interactions, antibonding NN and FeFe interactions exist in the crystal structure.
Co-reporter:Volker L. Deringer, Andrei L. Tchougréeff, and Richard Dronskowski
The Journal of Physical Chemistry A 2011 Volume 115(Issue 21) pp:5461-5466
Publication Date(Web):May 6, 2011
DOI:10.1021/jp202489s
Simple, yet predictive bonding models are essential achievements of chemistry. In the solid state, in particular, they often appear in the form of visual bonding indicators. Because the latter require the crystal orbitals to be constructed from local basis sets, the application of the most popular density-functional theory codes (namely, those based on plane waves and pseudopotentials) appears as being ill-fitted to retrieve the chemical bonding information. In this paper, we describe a way to re-extract Hamilton-weighted populations from plane-wave electronic-structure calculations to develop a tool analogous to the familiar crystal orbital Hamilton population (COHP) method. We derive the new technique, dubbed “projected COHP” (pCOHP), and demonstrate its viability using examples of covalent, ionic, and metallic crystals (diamond, GaAs, CsCl, and Na). For the first time, this chemical bonding information is directly extracted from the results of plane-wave calculations.
Co-reporter:Michael Wessel
Journal of the American Chemical Society 2010 Volume 132(Issue 7) pp:2421-2429
Publication Date(Web):February 2, 2010
DOI:10.1021/ja910570t
The nature of nitrogen−nitrogen bonding and the metal oxidation states within late-noble-metal pernitrides have been determined by a series of density-functional electronic-structure calculations. In contrast to alkaline-earth pernitrides such as BaN2 which contain quasi-molecular double-bonded N22− units, compounds such as PtN2 incorporate a tetravalent metal and a N24− species with a N−N single bond due to four surplus electrons within the antibonding 1πg* molecular orbital. This fact is the source of the huge bulk moduli of PtN2 and related materials such as OsN2 and IrN2. The crystal structure of lanthanum pernitride, LaN2 ⇄ La3+ + N22− + e−, yet to be made, has been predicted, and its electronic structure is compared with a likewise hypothetical LaN2 which consists of both N22− and N24− pernitride units together with a trivalent lanthanum cation. Finite-temperature DFT calculations predict a very moderate reaction pressure toward LaN2 starting from LaN and elemental nitrogen of less than 2 GPa at 300 K.
Co-reporter:Michael Krings ; Michael Wessel ; Wolfgang Wilsmann ; Paul Müller
Inorganic Chemistry 2010 Volume 49(Issue 5) pp:2267-2272
Publication Date(Web):February 2, 2010
DOI:10.1021/ic902065q
White powdery β-SrNCN was obtained by the solid-state metathesis between SrI2 and ZnNCN at 843 K, by the reaction of SrI2, CsCN, and CsN3 in tantalum cylinders at the same temperature, and from the direct reaction between elemental Sr and H2NCN dissolved in liquid ammonia. The solid-state reactions carried out at a higher 973 K yield white α-SrNCN. Both experimental data (X-ray diffraction (XRD), infrared spectroscopy, differential scanning calorimetry (DSC)) as well as GGA density-functional phonon calculations show that the β-phase is thermochemically more stable, by a minute 2 kJ/mol (electronic-structure theory) and about 6 kJ/mol (DSC), whereas the α-phase is slightly more dense. In addition, both XRD and DSC measurements reveal two distinct (endothermic) steps for the β-to-α phase transition, that is, first around 920 ± 20 K, then at 985 ± 15 K based on the X-ray data. Thermochemically, the upper heat effect is larger by a factor of 20.
Co-reporter:Xiaohui Liu ; Manfred Speldrich ; Paul Kögerler ; Richard Dronskowski ;Andrei L. Tchougréeff
Inorganic Chemistry 2010 Volume 49(Issue 16) pp:7414-7423
Publication Date(Web):July 23, 2010
DOI:10.1021/ic100599z
A new series of coordination-network compounds containing Ni(CN)2 and MX (M = Rb, Cs; X = Cl, Br) quasi two-dimensional sheets has been synthesized and structurally characterized. The tetragonal crystal structure (I4/mmm, no. 139) can be derived from the distorted perovskite type. Chemically, the Lewis-acidic Ni(CN)2 moieties accept halide ligands, resulting both in slightly elongated [Ni(NC)4X2]4− octahedra and strongly elongated [Ni(CN)4X2]4− octahedra that are charge-balanced by M+ cations. Quantum-chemical calculations of the effective Hamiltonian crystal field (EHCF) type indicate the simultaneous presence of high- and low-spin Ni2+ in a 1:1 ratio, in agreement with GGA+U and UV studies reported here. Our magnetic susceptibility data also corroborate the theoretical findings. An analysis of the magnetic superexchange paths in the new series of compounds is performed as well as that of the tentative magnetic state of the series.
Co-reporter:Jens Burghaus ; Michael Wessel ; Andreas Houben
Inorganic Chemistry 2010 Volume 49(Issue 21) pp:10148-10155
Publication Date(Web):October 1, 2010
DOI:10.1021/ic1016033
The recently published two-step ammonolysis reaction giving access to phase-pure GaFe3N has been reinvestigated. Thermochemical calculations show that a high-temperature route is necessary to avoid the formation of the competing GaN phase. Compared to the prior study showing a Vegard-like behavior (that is, a linear correlation between lattice parameter and elemental composition), improved X-ray analysis using Mo Kα1 radiation in combination with density-functional theory calculations reveal a more complicated behavior of the lattice parameter within the entire GaxFe4−xN series. The new finding originates from the magnetic properties, and the change in the magnetic ordering with increasing Ga content from ferromagnetic γ′-Fe4N to antiferromagnetically ordered GaFe3N, as observed from susceptibility measurements, is reproduced by different theoretical spin-alignment models, that is, a systematic evaluation of several antiferromagnetic spin orientations. Nonetheless, all structural models are based on the favored atomic ordering for GaFe3N, explainable by the strong affinity between iron and nitrogen.
Co-reporter:Bengt Hallstedt, Dejan Djurovic, Jörg von Appen, Richard Dronskowski, Alexey Dick, Fritz Körmann, Tilmann Hickel, Jörg Neugebauer
Calphad 2010 Volume 34(Issue 1) pp:129-133
Publication Date(Web):March 2010
DOI:10.1016/j.calphad.2010.01.004
Cementite (Fe3C) is one of the most common phases in steel. In spite of its importance, thermodynamic investigations, either experimental or theoretical, of cementite are infrequent. In the present work, the thermodynamic properties of cementite are reevaluated and Gibbs energy functions valid from 0 K upwards presented. At high temperature (1000 K and above), the Gibbs energy is practically unchanged compared to previous evaluations. The energy of formation at 0 K was also calculated using density functional theory. This energy of formation (+8 kJ/mol at 0 K) is in reasonable agreement with the present thermodynamic evaluation (+23.5 kJ/mol at 0 K and +27.0 kJ/mol at 298.15 K) and with a solution calorimetric measurement of the enthalpy of formation (+18.8 kJ/mol at 298.15 K). In addition, the heat capacity was calculated theoretically using ab initio data combined with statistical concepts such as the quasiharmonic approximation. The theoretical calculation agrees equally well as the present evaluation with experimental data, but suggests a different weighting of the experimental data. In order to use it directly in the thermodynamic evaluation further modifications in the Fe–C system, primarily of the fcc phase, would be required in order to reproduce phase equilibrium data with sufficient accuracy.
Co-reporter:Xiaojuan Tang;Hongping Xiang Dr.;Xiaohui Liu Dr.;Manfred Speldrich Dr. Dr.
Angewandte Chemie International Edition 2010 Volume 49( Issue 28) pp:4738-4742
Publication Date(Web):
DOI:10.1002/anie.201000387
Co-reporter:RalfPeter Stoffel;Claudia Wessel;Marck-Willem Lumey Dr.
Angewandte Chemie 2010 Volume 122( Issue 31) pp:5370-5395
Publication Date(Web):
DOI:10.1002/ange.200906780
Abstract
In diesem Aufsatz stellen wir einen auf elektronentheoretischen Rechnungen beruhenden Zugang zu einer quantenchemischen Thermochemie von Feststoffen vor. Zunächst gehen wir auf lokale und kollektive Atomauslenkungen ein und erläutern kurz den theoretischen Hintergrund. Die fundamentale Bedeutung der Phononen, ihre Dispersionsrelationen, ihre experimentelle Bestimmung und ihre Berechnung wird beleuchtet, gefolgt von der systematischen Konstruktion thermodynamischer Potentiale auf dieser Basis. Anschließend liefern wir eine Rechenanleitung sowie eine kritische Analyse der erzielbaren Genauigkeit und zeigen dann, wie verschiedene festkörperchemische Probleme so angegangen werden können. Dazu gehört die Berechnung atomarer Aktivierungsenergien in perowskitischen Oxiden, aber auch die Berücksichtigung berechenbarer Schwingungsfrequenzen zur Aufklärung von Kristallstrukturen. Dann behandeln wir die klassisch oft beschriebene Druck- und Temperaturpolymorphie von elementarem Zinn und nehmen eine energetische Klassifikation metastabiler Oxidnitride des Tantals vor. Schließlich weisen wir für Hochtemperatursupraleiter nach, wie die Rechnungen zur Evaluation unbefriedigend präziser thermochemischer Daten herangezogen werden können. Am Ende zeigen wir die momentanen Grenzen und die zukünftigen Herausforderungen der Theorie auf.
Co-reporter:Hongping Xiang, Richard Dronskowski, Bernhard Eck, and Andrei L. Tchougréeff
The Journal of Physical Chemistry A 2010 Volume 114(Issue 46) pp:12345-12352
Publication Date(Web):November 1, 2010
DOI:10.1021/jp1081033
The electronic structures and magnetic properties of MNCN (M = Fe, Co, and Ni) have been investigated by density-functional theory including explicit electronic correlation through an ad hoc Coulomb potential (GGA+U). The results evidence CoNCN and NiNCN as type-II anti-ferromagnetic semiconductors (that is, intralayer ferromagnetic and interlayer anti-ferromagnetic), in accordance with experimental observations. Just like the prototype MnNCN, the MNCN phases, with M = Ni and Co, thus resemble the corresponding MO monoxides with respect to their magnetic and transport properties. By contrast, FeNCN remains (semi)metallic even upon applying a strong Coulomb correlation potential. This, most probably, is in contradiction with its observed optical transparency and expected insulating behavior and points toward a serious density-functional theory problem.
Co-reporter:RalfPeter Stoffel;Claudia Wessel;Marck-Willem Lumey Dr.
Angewandte Chemie International Edition 2010 Volume 49( Issue 31) pp:5242-5266
Publication Date(Web):
DOI:10.1002/anie.200906780
Abstract
In this contribution we introduce an electronic-structure-theory-based approach to a quantum-chemical thermochemistry of solids. We first deal with local and collective atomic displacements and explain how to calculate these. The fundamental importance of the phonons, their dispersion relations, their experimental determination as well as their calculation is elucidated, followed by the systematic construction of the thermodynamic potentials on this basis. Subsequently, we provide an introduction for practical computation as well as a critical analysis of the level of accuracy obtainable. We then show how different solid-state chemistry problems can be solved using this approach. Among these are the calculation of activation energies in perovskite-like oxides, but we also consider the use of theoretical vibrational frequencies for determining crystal structures. The pressure and temperature polymorphism of elemental tin which has often been classically described is also treated, and we energetically classify the metastable oxynitrides of tantalum. We also demonstrate, using the case of high-temperature superconductors, that such calculations may be used for an independent evaluation of thermochemical data of unsatisfactory accuracy. Finally, we show the present limits and the future challenges of the theory.
Co-reporter:Xiaojuan Tang;Hongping Xiang Dr.;Xiaohui Liu Dr.;Manfred Speldrich Dr. Dr.
Angewandte Chemie 2010 Volume 122( Issue 28) pp:4846-4850
Publication Date(Web):
DOI:10.1002/ange.201000387
Co-reporter:Andreas Houben, Vladimir Šepelák, Klaus-Dieter Becker and Richard Dronskowski
Chemistry of Materials 2009 Volume 21(Issue 5) pp:784
Publication Date(Web):February 4, 2009
DOI:10.1021/cm803004v
A novel two-step route for the synthesis of RhFe3N is presented that yields the target material with a significantly improved phase purity. Except from Fe0.5Rh0.5, the absence of other side products is exemplified on the basis of powder XRD, REM/EDX measurements, and Mössbauer investigations. As predicted from density-functional theory calculations, both Rietveld refinement and Mössbauer analysis show that the Rh atom within RhFe3N exclusively occupies Wyckoff position 1a but not 3c of the anti-perovskite-like structure in space group Pm3̅m, and the rhodium occupation of the 1a site is around 80%, yielding a precise formula of 1a(Rh0.8Fe0.2)3c(Fe3)1bN. The decomposition temperature of the nitride is estimated from temperature-dependent X-ray measurements to lie around 530 °C.
Co-reporter:Andreas Houben, Jens Burghaus and Richard Dronskowski
Chemistry of Materials 2009 Volume 21(Issue 18) pp:4332
Publication Date(Web):August 26, 2009
DOI:10.1021/cm901864z
The recently published two-step ammonolysis was adapted to the first synthesis of GaFe3N as a pure phase and AlFe3N with a very high phase purity. Similar to archetypal γ′-Fe4N (a = 3.7900(6) Å), the ternary nitride GaFe3N adopts a perovskite-like structure in space group Pm3̅m with a slightly enlarged lattice parameter of a = 3.7974(1) Å. The magnetic characterization clearly evidences that GaFe3N is an antiferromagnet. The systematic exchange of Ga by Fe in going from GaFe3N to γ′-Fe4N results in a change from antiferromagnetic to ferromagnetic behavior. In contrast, the homologous ternary nitride AlFe3N shows a statistical Al/Fe substitution which results in space group Fm3̅m. AlFe3N can be classified as a soft ferromagnetic nitride.
Co-reporter:Jens Burghaus, Richard Dronskowski, Gordon J. Miller
Journal of Solid State Chemistry 2009 Volume 182(Issue 10) pp:2613-2619
Publication Date(Web):October 2009
DOI:10.1016/j.jssc.2009.07.017
First-principles, density-functional studies of several intermetallic borides of the general type M2M′Ru5−nRhnB2 (n=0–5; M=Sc, Ti, Nb; M′=Fe, Co) show that the variation in saturation magnetic moment with valence-electron count follows a Slater–Pauling curve, with a maximum moment occurring typically at 66 valence electrons. The magnetic moments in these compounds occur primarily from the 3d electrons of the magnetically active M′ sites, with some contribution from the Ru/Rh sites via magnetic polarization. Electronic DOS curves reveal that a rigid-band approach is a reasonable approximation for the estimation of saturation moments and the analysis of orbital interactions in this family of complex borides. COHP analyses of the M′−M′ orbital interactions indicate optimized interactions in the minority spin states for Co-containing phases, but strong bonding interactions remaining in Fe-containing phases.Theoretically determined (spin-polarized LMTO-GGA) local magnetic moments as a function of the chemical valence Z for various intermetallic borides.
Co-reporter:Hongping Xiang, Xiaohui Liu and Richard Dronskowski
The Journal of Physical Chemistry C 2009 Volume 113(Issue 43) pp:18891-18896
Publication Date(Web):October 7, 2009
DOI:10.1021/jp907458f
The electronic structure and magnetic properties of CuNCN have been reinvestigated by density-functional theory including explicit electronic correlation (GGA+U). The calculated results show that CuNCN is a semiconductor with a band gap of 0.1 eV and without any local magnetic moments, in practically quantitative agreement with experiment. The coupling constant (J1, J2, and J3) between the nearest-neighbor Cu(II) ions have been calculated as a function of the dihedral angle between the central CuN4 unit and its N═C═N carbodiimide ligands changing from 145° (experimental value) to 180° (hypothetical value). Only if the dihedral angle is close to 180°, CuNCN exhibits a local magnetic moment, characteristic for Cu2+, and adopts an antiferromagnetic structure (intralayer ferromagnetic, interlayer antiferro-magnetic). The former discrepancy between experiment and density functional theory appears to be solved.
Co-reporter:Takahiro Yamada Dr.;Xiaohui Liu Dr.;Ulli Englert Dr.;Hisanori Yamane Dr. Dr.
Chemistry - A European Journal 2009 Volume 15( Issue 23) pp:5651-5655
Publication Date(Web):
DOI:10.1002/chem.200900508
Co-reporter:Xiaohui Liu Dr.;Ludwig Stork;Manfred Speldrich Dr.;Heiko Lueken Dr. Dr.
Chemistry - A European Journal 2009 Volume 15( Issue 7) pp:1558-1561
Publication Date(Web):
DOI:10.1002/chem.200802422
Co-reporter:Thomas Fickenscher, Sudhindra Rayaprol, Jörg von Appen, Richard Dronskowski, Rainer Pöttgen, Kazimierz Łat̀ka and Jacek Gurgul
Chemistry of Materials 2008 Volume 20(Issue 4) pp:1381
Publication Date(Web):January 31, 2008
DOI:10.1021/cm7020406
The silicide carbide GdRu2SiC was synthesized by arc-melting from the Laves phase GdRu2, silicon, and graphite. GdRu2SiC was characterized via X-ray powder and single-crystal data: DyFe2SiC type, Cmcm, a = 383.0(1) pm, b = 1106.9(2) pm, c = 715.7(1) pm, wR2 = 0.0363, 626 F2 values, and 20 variables. The silicon atoms have a distorted trigonal prismatic Gd2Ru4 coordination and the carbon atoms fill compressed [Gd4Ru2] octahedra. The shortest interatomic distances and strongest bondings occur for the Ru−C (187 pm) and Ru−Si (247 pm) contacts, the former ones even exhibiting double-bond strength. Together, these atoms build up a three-dimensional [Ru2SiC] network in which the gadolinium atoms fill channels. The magnetic and electronic properties of GdRu2SiC have been investigated by means of magnetometric and 155Gd Mössbauer spectroscopy measurements. Magnetic susceptibility measurements exhibit magnetic ordering with a broad feature around 10 K. Susceptibility increases below the broad peak, indicating complex magnetism in this compound. This fact is supported by Mössbauer spectroscopy, which exhibits a dramatic change of the Mössbauer spectrum below 4.2 K, indicating another magnetic phase transition. The results of Mössbauer spectroscopy are discussed here in detail to understand the nature of the magnetic ordering in GdRu2SiC below and above 4.2 K.
Co-reporter:Holger Wolff, Martin Lerch, Heikko Schilling, Carsten Bähtz, Richard Dronskowski
Journal of Solid State Chemistry 2008 Volume 181(Issue 10) pp:2684-2689
Publication Date(Web):October 2008
DOI:10.1016/j.jssc.2008.06.044
Magnesium-doped tantalum oxynitrides, which were prepared by ammonolysis of amorphous mixed oxides, have been investigated using quantum-theoretical methods. For small magnesium amounts (5 cat%), density-functional total-energy calculations indicate an anatase-type structure consisting of stretched, corner-sharing TaO3N3TaO3N3 octahedra with a tetrahedrally distorted equatorial plane. The calculated structural parameters are in excellent agreement with those obtained using X-ray powder diffraction and synchrotron radiation. Additionally, the quantum-chemical results show a clear preference for an ordered anionic distribution (space group I41mdI41md, no. 109) of the host lattice, which is locally disturbed around Mg2+Mg2+. For thermodynamical reasons, the excess oxygen anions, which replace nitrogen on account of the lower charge of the dopant cation, segregate next to magnesium, thus forming local MgO “domains”. For higher magnesium contents (⩾10%)(⩾10%), minor phases of rutile-type structure have to be expected, which is in good agreement with experimental data.Density-functional total energy of Mg-doped TaON in several polymorphs, each in its most stable arrangement, as a function of the dopant amount.
Co-reporter:Manuel Krott, Xiaohui Liu, Paul Müller, Richard Dronskowski
Journal of Solid State Chemistry 2007 Volume 180(Issue 1) pp:307-312
Publication Date(Web):January 2007
DOI:10.1016/j.jssc.2006.10.021
Well-crystallized cobalt and nickel hydrogencyanamide, Co(HNCN)2 and Ni(HNCN)2, were synthesized from the corresponding ammonia complexes [M(NH3)6]2+ under aqueous cyanamide conditions. The X-ray and neutron powder data evidence the orthorhombic system and space group Pnnm. The cell parameters for Co(HNCN)2 are a=6.572(1), b=8.805(2), c=3.267(1) Å, and Z=2; for the isotypic Ni(HNCN)2, the cell parameters arrive at a=6.457(1), b=8.768(2), c=3.230(1) Å. The octahedral coordinations of the metal ions are marginally squeezed, with interatomic distances of 4×Co–N=2.134(5) Å, 2×Co–N=2.122(9) Å, and 4×Ni–N=2.133(6) Å, 2×Ni–N=2.035(11) Å. The HNCN− units appear as slightly bent (177.5(2)° for Co(HNCN)2 and 175.7(2)° for Ni(HNCN)2) and exhibit cyanamide shape character due to triple- and single-bond C–N distances (1.20(2) vs. 1.33(2) Å for Co(HNCN)2 and 1.15(2) vs. 1.38(2) Å for Ni(HNCN)2). The infrared vibration data compare well with those of the three existing alkali-metal hydrogencyanamides.Crystal structure of Co(HNCN)2 and Ni(HNCN)2 with Co/Ni atoms as white, N as grey, C as dark-grey, and H as light-grey balls.
Co-reporter:Boniface P. T. Fokwa, Jörg von Appen and Richard Dronskowski
Chemical Communications 2006 (Issue 42) pp:4419-4421
Publication Date(Web):12 Sep 2006
DOI:10.1039/B608903H
The newly synthesized boride Ti1+xOs2−xRuB2 (x = 0.6) has a novel structure featuring one-dimensional chains of titanium atoms, one-dimensional strings of face-sharing empty tetrahedral and square pyramidal clusters and, most importantly, trigonal planar and strongly bonded B4 units with a B–B distance of 1.89 Å.
Co-reporter:Wuping Liao;Ulli Englert
European Journal of Inorganic Chemistry 2006 Volume 2006(Issue 21) pp:
Publication Date(Web):14 SEP 2006
DOI:10.1002/ejic.200600540
Eu2I2(NCN) is the first compound to contain discrete empty europium tetrahedra and infinite metal chains that are constructed from edge-sharing Eu6 units with an open handbag-like motif. The europium tetrahedra and chains are bridged by coordinating carbodiimide (NCN2–) anions in a three-dimensional network. (© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2006)
Co-reporter:H. Wolff, H. Schilling, M. Lerch, R. Dronskowski
Journal of Solid State Chemistry 2006 Volume 179(Issue 8) pp:2265-2270
Publication Date(Web):August 2006
DOI:10.1016/j.jssc.2006.01.055
Fluorite-type phases in the system Y–Ta–O–N have been studied using both first-principle electronic-structure calculations and molecular-dynamic simulations to validate the structural data and to explain unusual asymmetric reflection profiles observed in the experimental X-ray diffraction patterns. We provide evidence that the compounds may be macroscopically described as to represent cubic fluorite-type defect structures despite the fact that DFT calculations clearly show that all crystallographic unit cells appear as triclinically distorted. Additionally, we find that there is a minute (but hardly significant) tendency for anionic ordering at absolute zero temperature but none under reaction conditions.Structural result of a room-temperature molecular-dynamic simulation of a supercell of Y0.125Ta0.875O0.875N□0.125.
Co-reporter:Jörg von Appen;Marck-Willem Lumey Dr. Dr.
Angewandte Chemie 2006 Volume 118(Issue 26) pp:
Publication Date(Web):31 MAY 2006
DOI:10.1002/ange.200600431
Falsifiziert: Die Hochdrucksynthese von Platinnitrid, PtN, und seine vorgeschlagene Struktur werden durch voraussetzungsfreie Elektronenstrukturrechnungen widerlegt. Thermochemische Argumente beweisen, dass PtN im wenig dichten Zinkblendetyp sowohl energetisch äußerst instabil als auch unmöglich unter Hochdruck zu erhalten ist. Hingegen ist unter anderem der viel dichtere Cooperit-Typ (siehe Bild) ein viel aussichtsreicherer Kandidat für die 1:1-Zusammensetzung.
Co-reporter:Jörg von Appen;Marck-Willem Lumey Dr. Dr.
Angewandte Chemie International Edition 2006 Volume 45(Issue 26) pp:
Publication Date(Web):31 MAY 2006
DOI:10.1002/anie.200600431
Mistaken identity: The high-pressure synthesis of PtN in the zinc blende structure type is refuted on the basis of first-principles electronic-structure calculations, which demonstrate that this low-density phase is energetically unstable and cannot be produced at high pressure. The denser cooperite structure type is preferable for PtN (see picture; NPt4 tetrahedra are yellow).
Co-reporter:Wuping Liao, Boniface P. T. Fokwa and Richard Dronskowski
Chemical Communications 2005 (Issue 28) pp:3612-3614
Publication Date(Web):09 Jun 2005
DOI:10.1039/B504235F
Eu8(NCN)4.95I6.10 is the first compound with discrete tritetrahedral Eu8 clusters which are interconnected by coordinating NCN2− carbodiimide anions on their triangular faces to form separated layers, the latter being bridged by iodide and carbodiimide anions.
Co-reporter:Andreas Houben, Paul Müller, Jörg von Appen, Heiko Lueken, Rainer Niewa,Richard Dronskowski
Angewandte Chemie International Edition 2005 44(44) pp:7212-7215
Publication Date(Web):
DOI:10.1002/anie.200502579
Co-reporter:Andreas Houben Dipl.-Chem.;Paul Müller Dr.;Jörg von Appen Dipl.-Chem.;Heiko Lueken Dr.;Rainer Niewa Dr. Dr.
Angewandte Chemie 2005 Volume 117(Issue 44) pp:
Publication Date(Web):18 OCT 2005
DOI:10.1002/ange.200502579
Vorhersage bestätigt: Die kürzlich durch Gesamtenergierechnungen vorhergesagte Phase RhFe3N (siehe Elementarzelle) mit außergewöhnlichen magnetischen Eigenschaften wurde erstmals synthetisiert. Der experimentelle Gitterparameter stimmt mit dem berechneten Wert gut überein, und die Phase entpuppt sich als halbharter itineranter Ferromagnet. Das atomare Sättigungsmoment beträgt =8.3 μB pro Formeleinheit, die Curie-Temperatur TC=505(25) K.
Co-reporter:Jörg von Appen
Angewandte Chemie International Edition 2005 Volume 44(Issue 8) pp:
Publication Date(Web):5 JAN 2005
DOI:10.1002/anie.200462247
Cutting corners: The perovskite-like ternary nitrides MFe3N of the iron- and platinum-group metals are theoretically investigated. All eight compounds are predicted as being ferromagnetic, with saturation moments between 7.1 and 9.2 μB per formula unit, and with larger M atoms preferentially occupying the corner position 1a (see picture). Two ternary nitrides, yet to be made, are predicted as lucrative synthetic goals.
Co-reporter:Jörg von Appen Dipl.-Chem.
Angewandte Chemie 2005 Volume 117(Issue 8) pp:
Publication Date(Web):5 JAN 2005
DOI:10.1002/ange.200462247
Die perowskitähnlichen ternären Nitride MFe3N der Metalle der Eisen- und Platingruppe wurden theoretisch untersucht. Alle acht Verbindungen werden als ferromagnetisch vorhergesagt, mit Sättigungsmomenten zwischen 7.1 und 9.2 μB pro Formeleinheit, und insbesondere größere M-Atome nehmen bevorzugt die Eckenposition 1a ein (siehe Bild). Zwei noch nicht synthetisierte ternäre Nitride werden als lohnende Syntheseziele ausgemacht.
Co-reporter:M.-W. Lumey;R. Dronskowski
Advanced Functional Materials 2004 Volume 14(Issue 4) pp:
Publication Date(Web):17 MAR 2004
DOI:10.1002/adfm.200305090
We report electronic structure calculations using density-functional theory (local density approximation (LDA) and generalized gradient approximation (GGA); plane waves and muffin-tin orbitals; pseudopotentials and all-electron approaches) on non-stoichiometric CoNxO1–x oxynitride phases. The preference of the experimentally suggested zinc-blende structure type over the rock-salt type is confirmed and explained, on the basis of COHP (crystal orbital Hamilton population) chemical bonding analyses, by reduced Co–Co antibonding interactions in the ZnS structural alternative. A pressure-induced phase transition into the NaCl type, however, is predicted at approximately 30 GPa. Supercell calculations touching upon the exact composition and local structure of CoNxO1–x provide evidence for a broad range of energetically metastable compositions with respect to the zinc-blende-type boundary phases CoN and CoO, especially for the more oxygen-rich phases. All non-stoichiometric compounds are predicted to be metallic materials which do not exhibit significant magnetic moments. Likewise, there is no indication for anionic ordering such that random anion arrangements are preferred.
Co-reporter:Wuping Liao, Jörg von Appen and Richard Dronskowski
Chemical Communications 2004 (Issue 20) pp:2302-2303
Publication Date(Web):02 Sep 2004
DOI:10.1039/B408647C
LiSr2(NCN)I3, the first extended compound containing empty tetrahedral Sr4 entities, is synthesized using a new flux route, and it exhibits two interpenetrating three-dimensional networks made up from Sr tetrahedra capped by NCN2− anions on their triangular faces and vertex-sharing LiI6 octahedra.
Co-reporter:Philipp Kölle
European Journal of Inorganic Chemistry 2004 Volume 2004(Issue 14) pp:
Publication Date(Web):6 JUL 2004
DOI:10.1002/ejic.200400507
See original Eur. J. Inorg. Chem. 2004, 2313–2320 ( DOI: http://dx.doi.org/10.1002/ejic.200300940).
Co-reporter:Philipp Kölle
European Journal of Inorganic Chemistry 2004 Volume 2004(Issue 11) pp:
Publication Date(Web):7 APR 2004
DOI:10.1002/ejic.200300940
The crystal structures of three very low-melting ionic liquid (IL) salts containing the 1-butyl-2,3-dimethylimidazolium cation and the fluoro anions BF4−, PF6− and SbF6− are reported. These are considered model compounds for ambient temperature ionic liquids, which are currently being explored as solvents for various types of synthetic reactions. All three compounds adopt significantly different structures in the solid state, highlighting the influence of counterion size in a series comprising a common cation. The crystal structures are discussed with respect to the possible existence of polar and non-polar domains, which are considered to be involved with the solvation of polar and non-polar substrates. The melting points of all three compounds are within the range 40−45 °C. A comparative measurement of conductivities of the molten salts in the vicinity of 20−50 °C has been made, including the new IL 1-allyl-2,3-dimethylimidazolium tetrafluoroborate and the widely applied 1-butyl-3-methylimidazolium tetrafluoroborate (BMIMBF4). (© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2004)
Co-reporter:Jörg von Appen, Richard Dronskowski, Klaus Hack
Journal of Alloys and Compounds 2004 Volume 379(1–2) pp:110-116
Publication Date(Web):6 October 2004
DOI:10.1016/j.jallcom.2004.03.002
The binary system Sn/Zn was theoretically investigated by a classical thermodynamic analysis (CALPHAD approach) and by density-functional total-energy calculations on the basis of the LDA/GGA, plane waves/muffin-tin orbitals, and supercell geometries. In harmony with experimental data, both methods agree in that there is only very small solubility between the elements and no formation of a stable intermetallic phase over the entire compositional range. For the hypothetical composition Sn2Zn, a total of 30 different crystal structures was quantum-mechanically optimized, and the chemical bondings of Sn2Zn adopting the CaF2 and HgBr2 structures were analyzed in detail; generally, the more ionic structure types are better suited for the Sn2Zn composition than typical intermetallic ones. Theoretical enthalphy-pressure diagrams were generated to explore high-pressure compound formation, and the observed transition pressures between the α, β and γ allotropes of tin were correctly reproduced by electronic structure theory.
Co-reporter:Michael Scholten, Philipp Kölle, Richard Dronskowski
Journal of Solid State Chemistry 2003 Volume 174(Issue 2) pp:349-356
Publication Date(Web):September 2003
DOI:10.1016/S0022-4596(03)00243-3
The mixed-valent compound In4Br7 undergoes a higher-order phase transition below which leads to a decrease in symmetry from the trigonal to the monoclinic (C2/c) system via . The phase transition has been monitored by X-ray powder diffraction using a linear position-sensitive detector between 15 and , and the crystal structures at room temperature and at 90 K have been refined by means of time-of-flight neutron powder-diffraction data; at , the lattice parameters are , , , and β=98.20(1)°; the new unit cell contains 88 atoms (Z=8) of which 12 are symmetry-independent. Due to their electronic instability because of a second-order Jahn–Teller effect, two of the three crystallographically independent monovalent indium cations are severely affected by the phase transition with respect to their coordination spheres; bond-valence calculations reveal significant strengthening of In+–Br− bonding upon symmetry reduction. Structural changes and group–subgroup relationships as well as possible intermediate phases are discussed.
Co-reporter:Yasemin Kurtulus, Richard Dronskowski
Journal of Solid State Chemistry 2003 Volume 176(Issue 2) pp:390-399
Publication Date(Web):December 2003
DOI:10.1016/S0022-4596(03)00242-1
The electronic structure of ferromagnetic τ-MnAl has been calculated using density-functional techniques (TB-LMTO-ASA, FLAPW) and quantum-chemically analyzed by means of the crystal orbital Hamilton population tool. While all observable quantities are in good agreement with experiment, the tetragonal structure of ferromagnetic MnAl is interpreted to arise from a nonmagnetic cubic structure by two subsequent steps, namely (a) an electronic distortion due to spin polarization followed by (b) a structural distortion into the tetragonal system. The various strengths of interatomic bonding have been calculated in order to elucidate the competition between electronic and structural distortion.
Co-reporter:Xiaohui Liu Dr.;Paul Müller Dr.;Peter Kroll Dr. Dr.;Wolfgang Wilsmann Dr.;Reinhard Conradt Dr.
ChemPhysChem 2003 Volume 4(Issue 8) pp:
Publication Date(Web):7 AUG 2003
DOI:10.1002/cphc.200390094
Co-reporter:Richard Dronskowski Dr.;Karol Korczak;Heiko Lueken Dr.;Walter Jung Dr.
Angewandte Chemie International Edition 2002 Volume 41(Issue 14) pp:
Publication Date(Web):15 JUL 2002
DOI:10.1002/1521-3773(20020715)41:14<2528::AID-ANIE2528>3.0.CO;2-6
Quaternary rhodium borides of general formula A2MRh5B2 (see picture: Mg green, Mn red, Rh blue, B yellow) offer a nice playground for combined synthetic–theoretical investigations. The relative robustness of the underlying structure type allows various adjustments of the valence-electron concentration to be explored. In synthesizing new magnetic materials by following a chemical theory of cooperative magnetic phenomena, it is demonstrated how physical properties, such as antiferromagnetic or ferromagnetic behavior can be understood, predicted, and, eventually, realized.
Co-reporter:Richard Dronskowski Dr.;Karol Korczak;Heiko Lueken Dr.;Walter Jung Dr.
Angewandte Chemie 2002 Volume 114(Issue 14) pp:
Publication Date(Web):15 JUL 2002
DOI:10.1002/1521-3757(20020715)114:14<2638::AID-ANGE2638>3.0.CO;2-0
Quaternäre Rhodiumboride der allgemeinen Zusammensetzung A2MRh5B2 (z. B. Mg2MnRh5B2, siehe Bild; Mg grün, Mn rot, Rh blau, B gelb) sind eine wahre Spielwiese für kombinierte synthetisch-theoretische Untersuchungen, denn der vergleichsweise robuste Strukturtyp ermöglicht es, unterschiedliche Valenzelektronenkonzentrationen auszutesten. Es wurde gezeigt, dass eine gewünschte physikalische Eigenschaft eines magnetischen Materials, wie antiferromagnetisches oder ferromagnetisches Verhalten, verstanden, vorhergesagt und letztlich auch präparativ realisiert werden kann.
Co-reporter:Gregory A. Lrum Dr. and Dr.
Angewandte Chemie 2000 Volume 112(Issue 24) pp:
Publication Date(Web):15 DEC 2000
DOI:10.1002/1521-3757(20001215)112:24<4647::AID-ANGE4647>3.0.CO;2-N
Co-reporter:Gregory A. Lrum Dr. and Dr.
Angewandte Chemie 2000 Volume 112(Issue 9) pp:
Publication Date(Web):2 MAY 2000
DOI:10.1002/(SICI)1521-3757(20000502)112:9<1598::AID-ANGE1598>3.0.CO;2-Y
Es wird eine chemische Betrachtung spinmagnetischer Phänomene in abgeschlossenen (Atome und Moleküle) und unendlich großen Systemen (Übergangsmetalle und ihre Legierungen) vorgestellt, die auf den Konzepten von Bindung und elektronischer Abschirmung basiert. Der Ansatz soll als halbquantitative Richtschnur für die Synthese neuer Ferromagnete dienen. Nach einem gestrafften Überblick der historischen Entwicklung verwandter Theorien aus der Physik werden die Auswirkungen der Spin-Spin-Kopplung, wie sie sich im Austausch- oder Fermi-Loch manifestieren, für Atome und Moleküle aufgezeigt. Beim Übergang in den paramagnetischen Zustand werden die Majoritäts- und Minoritätsspins stärker bzw. schwächer an den Atomkern gebunden, und es resultieren Unterschiede für die Energien und Raumbedürfnisse der zwei Sätze an Spinorbitalen. Das nur spärliche Auftreten ferromagnetischer Übergangsmetalle ergibt sich, nach Extrapolation gängiger Argumente aus der Ligandenfeldtheorie, wegen der Unterdrückung des Paramagnetismus freier Atome aufgrund der starken interatomaren Wechselwirkungen im festen Zustand. Allerdings führen kritische Valenzelektronenkonzentrationen in Fe, Co und Ni wegen der Besetzung antibindender Zustände am Ferminiveau εF zu lokalen elektronischen Instabilitäten. Die Entfernung jener antibindenden Zustände aus der Nachbarschaft um εF ist der Ursprung des Ferromagnetismus; in den reinen Metallen führt dies zu einer Verstärkung der chemischen Bindung. In den 4d- und 5d-Übergangsmetallen sind die Valenz-d-Orbitale bereits zu stark vom Kern abgeschirmt, und deshalb führt der gedachte Übergang in den ferromagnetischen Zustand nicht zu genügend großen Energieänderungen. Insofern weist das außergewöhnliche Auftreten des Ferromagnetismus allein in der ersten Übergangsreihe Parallelen zur speziellen Hauptgruppenchemie der ersten Langperiode auf. Eine Verknüpfung des Ferromagnetismus der Übergangsmetalle mit Pearsons absoluter chemischer Härte η fällt leicht, und die Bedingung für das Auftreten des Ferromagnetismus in den nichtmagnetischen Rechnungen lautet η<0.2 eV. Wie man es vom Prinzip der maximalen Härte erwartet, werden Fe, Co und Ni beim Übergang in den stabileren, ferromagnetischen Zustand durchgängig härter. Der Magnetismus intermetallischer Legierungen ist wesensgleich. Unabhängig davon, ob eine Legierung ein ferromagnetisches Element enthält, bedeutet die Anwesenheit antibindender Zustände an εF einen „Fingerabdruck” für ferromagnetische Instabilität. Die Größenunterschiede der lokalen magnetischen Momente auf den konstituierenden Atomen einer ferromagnetischen Legierung können im Rahmen der relativen Beiträge zur Zustandsdichte an εF in den nichtmagnetischen Rechnungen verstanden werden. Sofern geschickt parametrisiert, erlauben auch nichtmagnetische, semiempirische Rechnungen das Aufzeigen ferromagnetischer Instabilitäten in Elementen und Legierungen. Darum können diese Techniken, die mittlerweile fast schon überall zu finden sind, den Synthetiker auf der Suche nach neuen ferromagnetischen Materialien anleiten.
Co-reporter:Gregory A. Lrum and
Angewandte Chemie 1999 Volume 111(Issue 10) pp:
Publication Date(Web):11 MAY 1999
DOI:10.1002/(SICI)1521-3757(19990517)111:10<1481::AID-ANGE1481>3.0.CO;2-3
Voraussetzungsfreie Bandstrukturrechnungen sind die Grundlage für eine einfache Erklärung des Auftretens von Ferromagnetismus in elementarem Eisen, Cobalt und Nickel, die auf einem entscheidenden chemischen Konzept beruht: Bindung. Es wird beschrieben, daß das Einsetzen des Ferromagnetismus die Metall-Metall-Bindungen in diesen Übergangsmetallen durch Abschwächung andernfalls antibindender Nächste-Nachbarn-Wechselwirkungen an der Fermi-Kante verstärkt.
Co-reporter:Gregory A. Lrum and
Angewandte Chemie International Edition 1999 Volume 38(Issue 10) pp:
Publication Date(Web):11 MAY 1999
DOI:10.1002/(SICI)1521-3773(19990517)38:10<1389::AID-ANIE1389>3.0.CO;2-K
Band structure calculations fromfirst principles provide the basis of a simple explanation for the appearance of ferromagnetism in iron, cobalt, and nickel that is founded upon a uniquely chemical concept: bonding. It is shown that the onset of ferromagnetism strengthens the metal–metal bonds in these transition metals by reducing the antibonding nearest neighbor interactions that would otherwise appear at the Fermi level.
Co-reporter:Gregory A. Lrum;Rainer Niewa;Francis J. DiSalvo
Chemistry - A European Journal 1999 Volume 5(Issue 2) pp:
Publication Date(Web):4 FEB 1999
DOI:10.1002/(SICI)1521-3765(19990201)5:2<515::AID-CHEM515>3.0.CO;2-Y
A systematic theoretical examination of the electronic structures of a number of Ce compounds reveals some qualitative trends in the band structures that may be useful in the assignment of Ce oxidation state: in Ce3+ compounds there is at least one occupied band which is clearly made up mostly of Ce states, while in systems containing Ce4+ the small Ce contributions to occupied states are spread across many bands. The figure shows an electron density contour plot of an energy slice of CeN.
Co-reporter:Volker L. Deringer, Ralf P. Stoffel, Matthias Wuttig and Richard Dronskowski
Chemical Science (2010-Present) 2015 - vol. 6(Issue 9) pp:NaN5262-5262
Publication Date(Web):2015/06/29
DOI:10.1039/C5SC00825E
Antimony selenide (antimonselite, Sb2Se3) is a versatile functional material with emerging applications in solar cells. It also provides an intriguing prototype to study different modes of bonding in solid chalcogenides, all within one crystal structure. In this study, we unravel the complex bonding nature of crystalline Sb2Se3 by using an orbital-based descriptor (the crystal orbital Hamilton population, COHP) and by analysing phonon properties and interatomic force constants. We find particularly interesting behaviour for the medium-range Sb⋯Se contacts, which still contribute significant stabilisation but are much softer than the “traditional” covalent bonds. These results have implications for the assembly of Sb2Se3 nanostructures, and bond-projected force constants appear as a useful microscopic descriptor for investigating a larger number of chalcogenide functional materials in the future.
Co-reporter:Marck Lumeij, Michael Gilleßen, Henny Bouwmeester, Torsten Markus, Juri Barthel, Stefan Roitsch, Joachim Mayer and Richard Dronskowski
Physical Chemistry Chemical Physics 2014 - vol. 16(Issue 4) pp:NaN1338-1338
Publication Date(Web):2013/11/20
DOI:10.1039/C3CP53958J
We present a theoretical and experimental study on the influence of the Ba/Sr and Co/Fe ratios as well as the oxygen-non-stoichiometry on the stability of Ba0.5Sr0.5Co0.8Fe0.2O3−δ (BSCF). Thin-layer depositions are analysed by looking at TEM images and EDX spectra. Bond-analytical calculations are performed to explain the stability difference between hexagonal and cubic BSCF. Finally, annealing experiments analysed using XRD give an insight into the differences of phase-fraction growth with respect to the Ba/Sr ratio.
Co-reporter:Volker L. Deringer, Wei Zhang, Pascal Rausch, Riccardo Mazzarello, Richard Dronskowski and Matthias Wuttig
Journal of Materials Chemistry A 2015 - vol. 3(Issue 37) pp:NaN9523-9523
Publication Date(Web):2015/08/26
DOI:10.1039/C5TC02314A
We identify a similar feature in the chemical-bonding nature of seemingly different phase-change materials (PCMs) for data storage. This affords new insight into the “next-generation” material In3SbTe2, establishes a hitherto missing link to the more ubiquitous Ge–Sb–Te alloys, and encourages the search for new PCMs beyond established electron-counting schemes.
Co-reporter:Peter Klaus Sawinski, Volker L. Deringer and Richard Dronskowski
Dalton Transactions 2013 - vol. 42(Issue 42) pp:NaN15087-15087
Publication Date(Web):2013/08/13
DOI:10.1039/C3DT51820E
A family of alkali-metal guanidinates (M = Na–Cs) has been reported recently, representing all-nitrogen analogues of alkali bicarbonates and containing the elusive guanidinate monoanion CN3H4−. Here, we describe the synthesis and characterisation of LiCN3H4 as the last representative of alkali guanidinates. Single crystals of LiCN3H4 were obtained using a solvothermal route in liquid ammonia, and the crystal structure was determined at 100 K using single-crystal X-ray diffraction (monoclinic, P21/c, Z = 4, a = 7.251(2) Å, b = 4.532(2) Å, c = 9.051(2) Å, β = 103.315(3)°). The structure of LiCN3H4 is reminiscent of the previously reported NaCN3H4 type in that both guanidinates contain tetrahedrally nitrogen-coordinated alkali-metal cations. The linking of these tetrahedra differs, however: they share corners in NaCN3H4 to form one-dimensionally infinite chains running through the crystal, whereas isolated, edge-sharing tetrahedra (Li2N6 motif) are found in the LiCN3H4 structure described here. Periodic density-functional theory (DFT) computations at the dispersion-corrected PBE + D2 level not only correctly reproduce the structural preferences, but also indicate that a NaCN3H4 polymorph should be viable adopting the lithium guanidinate type but much less easily so vice versa.
Co-reporter:Volker L. Deringer and Richard Dronskowski
Chemical Science (2010-Present) 2014 - vol. 5(Issue 3) pp:NaN903-903
Publication Date(Web):2013/11/15
DOI:10.1039/C3SC52743C
Germanium dioxide (GeO2) finds increasing application on the nanoscale, which calls for a detailed understanding of its crystal surfaces. In particular, the metastable α-quartz-type polymorph of GeO2 exhibits many desirable properties but also a non-trivial structural chemistry. Here, we contribute a surface study of quartz-type GeO2 in which we combine periodic density-functional theory (DFT) with classical chemical reasoning. We report on the most relevant surfaces, both freshly cleaved and structurally optimised. Stability trends of the latter are discussed in terms of the central structural unit—the [GeO4] tetrahedra—and how they are linked at the surface, in seamless extension of Pauling's third rule which had originally been conceived for bulk crystal structures. A more detailed, energy-resolved view is afforded by computing crystal orbital overlap populations (COOP) with a novel projection scheme; this way, a “bond strength” is directly gauged from plane-wave DFT output, and it allows the different surface stabilities to be rationalised in terms of “strengthened” and “weakened” bonds. These results and ways of thinking may be relevant for future studies on nanocrystalline GeO2 and, in a broader context, also for silica (SiO2) and other surfaces.
Co-reporter:Volker L. Deringer, Ulli Englert and Richard Dronskowski
Chemical Communications 2014 - vol. 50(Issue 78) pp:NaN11549-11549
Publication Date(Web):2014/08/08
DOI:10.1039/C4CC04716H
The covalent nature of short hydrogen bonds has been under debate for long. Here we show that the crystal orbital Hamilton population (COHP) bonding indicator gives new, complementary evidence of covalent hydrogen⋯acceptor interactions in the molecular solid state.
Co-reporter:T. Scholz and R. Dronskowski
Journal of Materials Chemistry A 2017 - vol. 5(Issue 1) pp:NaN175-175
Publication Date(Web):2016/11/22
DOI:10.1039/C6TC04543J
We present an experimental and theoretical study of the solid solution GexFe4−xNy (0 ≤ x ≤ 1). A two-step ammonolytic reaction gives access to the compounds with phase-pure quality. The GexFe4−xNy nitrides show a transition from an antiperovskite-like to a tetragonally distorted structure with increasing germanium concentration. Various experimental and theoretical methods evidence that the iron substitution by germanium exclusively takes place at the cubic Wyckoff position 1a. Despite the phase transition, one observes a Vegard-type decrease of the lattice parameter over the entire compositional range. In addition, the nitrides have a limited nitrogen capacity: incorporating germanium drastically reduces the nitrogen content in the cubic structure, but increases it again in the tetragonal structure. Combined HT-XRD and TG-DSC measurements evidence that the germanium-richest nitrides are highly expanding materials. They first show a transition to a cubic structure before they decompose by releasing nitrogen. Magnetic measurements reveal that the gradual germanium incorporation is accompanied by a drastic weakening of the ferromagnetic interactions leading to a frustrated iron spin system. Ge0.97Fe3.03N0.56 is identified as a canonical spin glass with the characteristic parameters Tg = 36.68(5) K, τ* = 10−13.8(2) s, zν = 7.2(1) and ΔTm/(Tm·Δlgω) = 0.012.























![Phenol, 4-[(4-aminophenyl)methyl]-](http://img.cochemist.com/ccimg/97600/97599-94-5.png)
![Phenol, 4-[(4-aminophenyl)methyl]-](http://img.cochemist.com/ccimg/97600/97599-94-5_b.png)






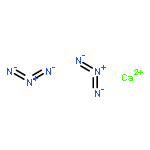

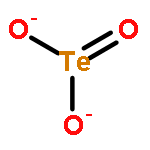
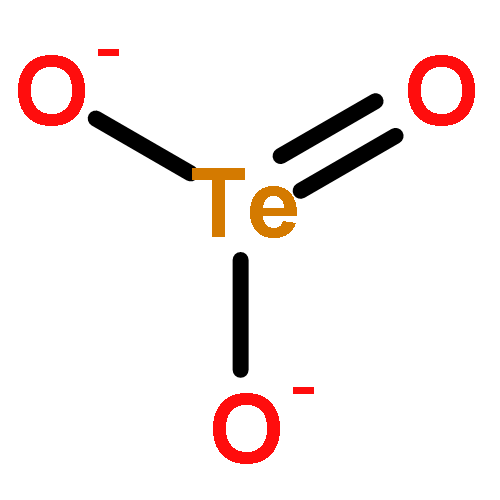







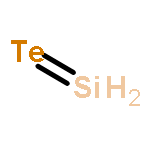
![Tetracyclo[3.2.0.02,7.04,6]heptane](http://img.cochemist.com/ccimg/300/278-06-8.png)
![Tetracyclo[3.2.0.02,7.04,6]heptane](http://img.cochemist.com/ccimg/300/278-06-8_b.png)




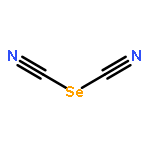
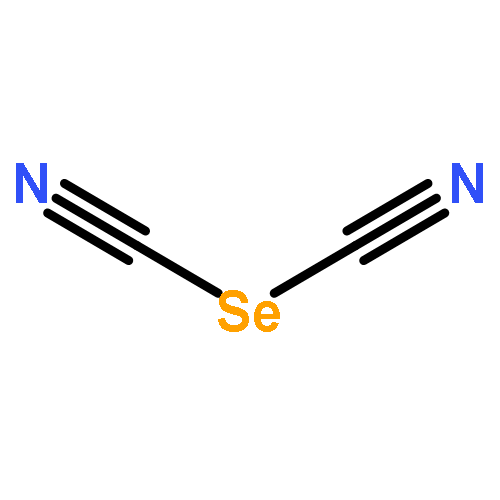
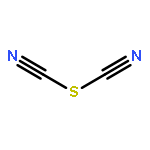
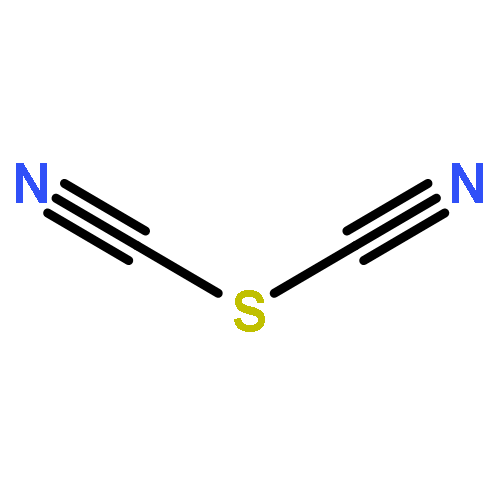

![[2-AZIDO-2-(2-PHENYL-4,5-DIHYDRO-1,3-OXAZOL-4-YL)ETHYL] 4-NITROBENZOATE](http://img.cochemist.com/ccimg/7300/7231-16-5.png)
![[2-AZIDO-2-(2-PHENYL-4,5-DIHYDRO-1,3-OXAZOL-4-YL)ETHYL] 4-NITROBENZOATE](http://img.cochemist.com/ccimg/7300/7231-16-5_b.png)