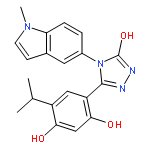
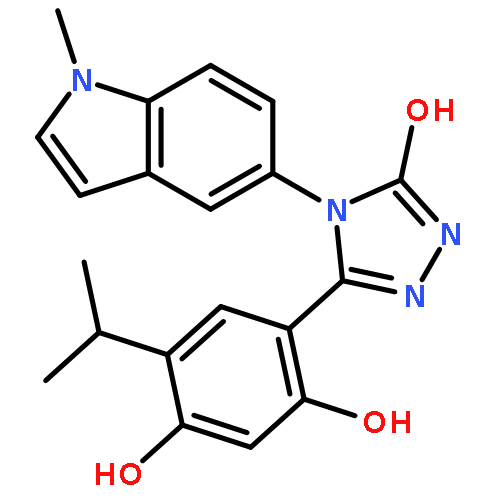
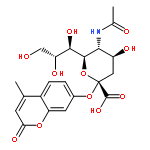
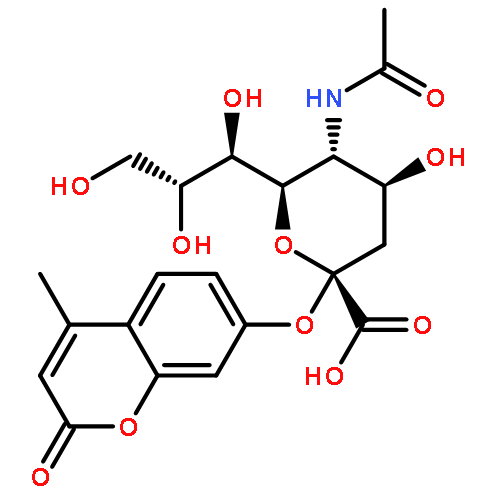
Co-reporter:Christopher L. Moore, Mahender B. Dewal, Emmanuel E. Nekongo, Sebasthian Santiago, Nancy B. Lu, Stuart S. Levine, and Matthew D. Shoulders
ACS Chemical Biology 2016 Volume 11(Issue 1) pp:200
Publication Date(Web):October 26, 2015
DOI:10.1021/acschembio.5b00740
Proteostasis in the cytosol is governed by the heat shock response. The master regulator of the heat shock response, heat shock factor 1 (HSF1), and key chaperones whose levels are HSF1-regulated have emerged as high-profile targets for therapeutic applications ranging from protein misfolding-related disorders to cancer. Nonetheless, a generally applicable methodology to selectively and potently inhibit endogenous HSF1 in a small molecule-dependent manner in disease model systems remains elusive. Also problematic, the administration of even highly selective chaperone inhibitors often has the side effect of activating HSF1 and thereby inducing a compensatory heat shock response. Herein, we report a ligand-regulatable, dominant negative version of HSF1 that addresses these issues. Our approach, which required engineering a new dominant negative HSF1 variant, permits dosable inhibition of endogenous HSF1 with a selective small molecule in cell-based model systems of interest. The methodology allows us to uncouple the pleiotropic effects of chaperone inhibitors and environmental toxins from the concomitantly induced compensatory heat shock response. Integration of our method with techniques to activate HSF1 enables the creation of cell lines in which the cytosolic proteostasis network can be up- or down-regulated by orthogonal small molecules. Selective, small molecule-mediated inhibition of HSF1 has distinctive implications for the proteostasis of both chaperone-dependent globular proteins and aggregation-prone intrinsically disordered proteins. Altogether, this work provides critical methods for continued exploration of the biological roles of HSF1 and the therapeutic potential of heat shock response modulation.
Co-reporter:Andrew S. DiChiara, Rebecca J. Taylor, Madeline Y. Wong, Ngoc-Duc Doan, Amanda M. Del Rosario, and Matthew D. Shoulders
ACS Chemical Biology 2016 Volume 11(Issue 5) pp:1408
Publication Date(Web):February 5, 2016
DOI:10.1021/acschembio.5b01083
Collagen-I is the most abundant protein in the human body, yet our understanding of how the endoplasmic reticulum regulates collagen-I proteostasis (folding, quality control, and secretion) remains immature. Of particular importance, interactomic studies to map the collagen-I proteostasis network have never been performed. Such studies would provide insight into mechanisms of collagen-I folding and misfolding in cells, an area that is particularly important owing to the prominence of the collagen misfolding-related diseases. Here, we overcome key roadblocks to progress in this area by generating stable fibrosarcoma cells that inducibly express properly folded and modified collagen-I strands tagged with distinctive antibody epitopes. Selective immunoprecipitation of collagen-I from these cells integrated with quantitative mass spectrometry-based proteomics permits the first mapping of the collagen-I proteostasis network. Biochemical validation of the resulting map leads to the assignment of numerous new players in collagen-I proteostasis, and the unanticipated discovery of apparent aspartyl-hydroxylation as a new post-translational modification in the N-propeptide of collagen-I. Furthermore, quantitative analyses reveal that Erp29, an abundant endoplasmic reticulum proteostasis machinery component with few known functions, plays a key role in collagen-I retention under ascorbate-deficient conditions. In summary, the work here provides fresh insights into the molecular mechanisms of collagen-I proteostasis, yielding a detailed roadmap for future investigations. Straightforward adaptations of the cellular platform developed will also enable hypothesis-driven, comparative research on the likely distinctive proteostasis mechanisms engaged by normal and disease-causing, misfolding collagen-I variants, potentially motivating new therapeutic strategies for currently incurable collagenopathies.
Co-reporter:Mahender B. Dewal, Andrew S. DiChiara, Aristotelis Antonopoulos, Rebecca J. Taylor, Chyleigh J. Harmon, Stuart M. Haslam, Anne Dell, Matthew D. Shoulders
Chemistry & Biology 2015 Volume 22(Issue 10) pp:1301-1312
Publication Date(Web):22 October 2015
DOI:10.1016/j.chembiol.2015.09.006
•XBP1s regulates N-glycan maturation pathways, in addition to proteostasis•XBP1s can enhance synthesis of hybrid and complex N-glycans on secreted proteins•Regulating N-glycodynamics is a new function for the unfolded protein response•XBP1s-mediated alterations in the N-glycome could have key biological impactsThe molecular architecture of the mature N-glycome is dynamic, with consequences for both normal and pathologic processes. Elucidating cellular mechanisms that modulate the N-linked glycome is, therefore, crucial. The unfolded protein response (UPR) is classically responsible for maintaining proteostasis in the secretory pathway by defining levels of chaperones and quality control proteins. Here, we employ chemical biology methods for UPR regulation to show that stress-independent activation of the UPR’s XBP1s transcription factor also induces a panel of N-glycan maturation-related enzymes. The downstream consequence is a distinctive shift toward specific hybrid and complex N-glycans on N-glycoproteins produced from XBP1s-activated cells, which we characterize by mass spectrometry. Pulse-chase studies attribute this shift specifically to altered N-glycan processing, rather than to changes in degradation or secretion rates. Our findings implicate XBP1s in a new role for N-glycoprotein biosynthesis, unveiling an important link between intracellular stress responses and the molecular architecture of extracellular N-glycoproteins.Figure optionsDownload full-size imageDownload high-quality image (184 K)Download as PowerPoint slide
Co-reporter:Angela M. Phillips, Matthew D. Shoulders
Journal of Molecular Biology (13 February 2016) Volume 428(Issue 3) pp:533-537
Publication Date(Web):13 February 2016
DOI:10.1016/j.jmb.2015.12.019
.jpg)