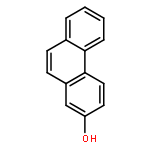
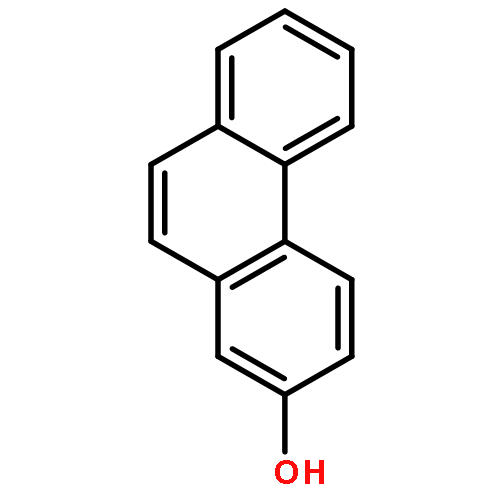
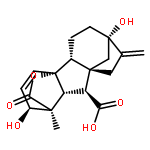
Co-reporter:Yi Yang, Shuo Yin, Yongxin Li, Dan Lu, Jing Zhang, Chengjun Sun
TrAC Trends in Analytical Chemistry 2017 Volume 95(Volume 95) pp:
Publication Date(Web):1 October 2017
DOI:10.1016/j.trac.2017.07.023
•Recent advances in aptamers-based detection methods of antibiotics in different matrices are reviewed.•Different aptasensors and aptamer-based purification methods are presented.•Current sensing strategies of antibiotics-detecting aptasensors are summarized.•Future trends of the application of aptamers in antibiotics detection are discussed.Antibiotic contamination and abuse are universal phenomena, accordingly antibiotic resistance has emerged as a serious issue globally in recent years. To detect antibiotics and their residues in the environment, foods, drugs and biological samples, numerous analytical techniques have been developed. Among them, aptamer-based methods are considered to be highly sensitive and selective and have been applied to the detection of various antibiotics in different samples. We present a systematical and critical review on the antibiotic-specific aptamers and their application in detection of antibiotics in different matrices, focusing on the recent advances in optical and electrochemical aptasensors, aptamer-affinity based sample purification, as well as the promising label-free and multiplex determination.
Co-reporter:Jing Zhang;Aimin Sun;Yi Yang;Jielan Hu;Ling Wei;Bo Gao;Xueqin Ding
Chromatographia 2016 Volume 79( Issue 23-24) pp:1649-1658
Publication Date(Web):2016 December
DOI:10.1007/s10337-016-3178-x
In this study, a method of field-amplified sample injection coupled with capillary zone electrophoresis with ultraviolet detection was established for evaluation of DNA methylation and hydroxymethylation levels in biological materials. By modifying an existing method, the separation of cytosine (C), 5-methylcytosine (5-mC) and 5-hydroxymethylcytosine (5-hmC) was performed on an uncoated capillary column (40 cm × 75 μm I.D.) using 300 mmol L−1 tris solution (pH 2.90) as running buffer and detected at 280 nm. The detection limits (S/N = 3) of the method were 0.004 ng mL−1 for cytosine (C), 0.01 ng mL−1 for 5-methylcytosine (5-mC), and 0.02 ng mL−1 for 5-hydroxymethylcytosine (5-hmC). The proposed method has been successfully applied to the evaluation of DNA methylation and hydroxymethylation levels of blood samples from 15 hepatocellular carcinoma patients and 5 liver cirrhosis patients and liver tissues from 50 pairs of tumor and matched tumor-adjacent samples.
Co-reporter:Yu Wang, Yongxin Li, Jinling Yang, Jia Ruan, Chengjun Sun
TrAC Trends in Analytical Chemistry 2016 Volume 78() pp:1-16
Publication Date(Web):April 2016
DOI:10.1016/j.trac.2015.08.010
•The MVOCs as indicators produced from different foodstuff were reviewed.•The sampling techniques for MVOCs from foodstuff were summarized.•The analytical methods for MVOCs from foodstuff were reviewed.•The approaches for identification of microbial genera by MVOCs were expounded.•The problems in the microorganism identification via MVOCs and future trends were explored.Foodborne diseases encompass a wide spectrum of illnesses and are an emerging public health concern worldwide. The contamination of food with microorganisms may occur at any stage in the process from food production to consumption. Multi-organ failure and even cancer may result from the ingestion of microorganism-contaminated foodstuffs, representing a considerable burden of disability as well as mortality. Thus, fast and effective approaches for evaluation of microbial contamination in foodstuff are vital. In recent years, the microbial volatile organic compounds (MVOCs), as indicators of microbial contamination, have already been playing an important role in clinic diagnosis and environment monitoring, and they have gradually being used to evaluate the microbial contamination in foodstuff. The microbial volatile organic compounds are a variety of volatile compounds produced by fungi, molds and bacteria during metabolism. By far, more than 1000 organic compounds have been identified as MVOCs comprised of alcohols, aldehydes, hydrocarbons, acids, ethers, esters, ketones, terpenoids, sulfur and nitrogen compounds. It is difficult to make reliable lists of MVOCs for relevant microbial species, since different microorganisms could share the same MVOCs. Although none of them can be considered to be exclusive or specific for certain microbial species, the identification of microbial species can be performed by statistical analysis of the MVOCs profiles. In addition, the approach based on enzyme-generated MVOCs specific to certain microbial species has also been applied in identification of microbial flora. In this paper, we summarize MVOCs in foodstuff and their application in food safety inspection. Moreover, the most commonly used analytical methods for detection of MVOCs are introduced as well. Additionally, the use of the approaches in the identification of microbial species is also expounded in this review.
Co-reporter:Li Zeng, Xin Wu, Yongxin Li, Dan Lu and Chengjun Sun
Analytical Methods 2015 vol. 7(Issue 2) pp:543-550
Publication Date(Web):10 Nov 2014
DOI:10.1039/C4AY02416H
A novel, simple and accurate high performance capillary electrophoresis method after multiwalled carbon nanotube-dispersive solid-phase extraction was developed for simultaneous determination of hydrochlorothiazide (HCT), chlortalidone (CTD), indapamide (IDP), reserpine (RSP), nifedipine (NDP) and valsartan (VST) in antihypertensive functional foods. After the analytes were ultrasonically extracted with acetonitrile, they were adsorbed on multiwalled carbon nanotubes (MWCNTs). Then the MWCNTs were separated through centrifugation and the analytes on the MWCNTs were desorbed with methanol. The eluent was removed through rotary evaporation and the residue was dissolved in acetonitrile–water (50:50, v/v) for CE analysis. The electrophoresis separation was carried out on an uncoated fused-silica capillary (57.0 cm total length and 50.0 cm effective length, 75.0 μm i.d.) by applying a voltage of 30 kV and the running buffer consisting of 10 mM borax buffer, 20 mM SDS and 30% acetonitrile (pH 9.7) with PDA detection at 220 nm. The capillary column temperature was set at 30 °C. The method showed good linearity in the ranges of 1–50 μg mL−1 with LODs of 0.058–0.157 μg mL−1. The proposed method was successfully applied to the analysis of antihypertensive functional foods with different matrices. Reserpine was detected in a sample with the content of 55.1 ± 0.9 μg mL−1 while other chemicals were not detected in all samples. The results of the proposed method were compared with those obtained by HPLC and there were no significant differences in the performance of the methods regarding accuracy and precision.
Co-reporter:Dan Lu, Yi Yang, Xin Wu, Li Zeng, Yongxin Li and Chengjun Sun
Analytical Methods 2015 vol. 7(Issue 8) pp:3353-3362
Publication Date(Web):03 Mar 2015
DOI:10.1039/C4AY02854F
A rapid and efficient gas chromatography-mass spectrometry (GC-MS) has been developed for determination of eight VE isomers including α-, β-, γ-, δ-tocopherols and tocotrienols, as well as α-tocopherol acetate in functional foods and nutritional supplements. The vitamin E isomers in samples were directly extracted without saponification with mixed solvents of methanol and hexane (7:3,v/v). Good separation was achieved using a VF-5MS column (30 m × 0.25 mm, 0.25 μm) within 13 min. The mass spectrometer was operated in both full scan mode and SIM mode using electron impact ionization. Qualitative detection was based on characteristic ion pairs and retention time. Dibenzanthracene was used as an internal standard for quantification measurement. The linear ranges of the method were from 0.1 to 40 μg mL−1 with the correlation coefficients greater than 0.997. The detection limits ranged from 0.09 ng mL−1 to 0.46 ng mL−1, and the quantification limits were from 0.29 ng mL−1 to 1.52 ng mL−1. The intra-day and inter-day relative standard deviations (RSDs) of the method (for 1 μg mL−1 standard solution) were in the range of 4.9% to 8.0% and 2.1% to 4.9%, respectively. The average recoveries of the method ranged from 83.2% to 107%, with the RSDs from 1.1% to 8.4%. The method has been applied for the determination of VE isomers and α-tocopherol acetate in functional foods and nutritional supplements with satisfactory results.
Co-reporter:Yi Yang;Dan Lu;Jing Zhang;Yongxin Li;Bo Zheng;Chengjun Sun
Chromatographia 2015 Volume 78( Issue 21-22) pp:1359-1366
Publication Date(Web):2015 November
DOI:10.1007/s10337-015-2951-6
An efficient, high-performance liquid-chromatographic method with diode-array detection (HPLC–DAD) has been established for simultaneous determination of retinol, α, (β + γ), and δ-tocopherols, and α, β, γ, and δ-tocotrienols in human serum. After deproteinization, the target vitamins in serum were extracted with n-hexane and the extract was evaporated under weak nitrogen flow. The residue was redissolved in methanol and the resulting solution was used for HPLC analysis. Retinol acetate and α-tocopherol acetate were used as internal standards. The internal standard calibration curves were linear over the range of 0.010–50.0 µg mL−1, with correlation coefficients >0.999. Mean recoveries of the method were 86.3–110 %, with intra-day and inter-day relative standard deviations less than 12.2 and 14.9 %, respectively. The detection limits of the method ranged from 0.001 to 0. 002 µg mL−1, and the quantification limits ranged from 0.002 to 0.008 µg mL−1. The method was successfully applied to analysis of the target vitamins in 50 human serum samples; all the analytes were detected at concentrations ranging from <0.002–23.0 µg mL−1.
Co-reporter:Luying Chen;Jiangtao Hu;Wei Zhang;Jing Zhang;Ping Guo
Food Analytical Methods 2015 Volume 8( Issue 8) pp:1903-1910
Publication Date(Web):2015 September
DOI:10.1007/s12161-014-0074-6
A high-performance capillary electrophoresis (HPCE) method for simultaneous determination of nine banned azo dyes including orange 2 sodium salt, ponceau SX, ponceau 2R, metanil yellow, yellow 2G, ponceau 3R, naphthol orange, acid violet 7, and acid yellow 11 in foodstuffs and beverages was established. After being extracted with methanol and cleaned up with Proelut PWA column, the separation of the target azo dyes in sample solution was performed in an uncoated fused-silica capillary (60 cm × 75 μm I.D.) with an effective length of 50 cm. The running buffer consisted of 15 % acetonitrile-10-mM borate buffer (pH = 9.0), and the applied voltage was 25 kV with the separation temperature of 25 °C. The calibration curves of the method were linear in the range of 0.50–100.0 μg/ml with correlation coefficients greater than 0.999. The recoveries of the method ranged from 81.2 to 110 %. The limits of detection (LODs) and the quantification of the method were in the ranges of 25.1–75.1 μg/l and 0.083–0.25 mg/kg, respectively. The relative standard deviations (RSDs) of interday and intraday were in the ranges of 1.90 to 5.81 % and 0.12 to 3.42 %, respectively. This method has been successfully applied to the simultaneous determination of aforementioned nine azo dyes in foodstuffs and beverages.
Co-reporter:Jinling Yang, Yongxin Li, Yu Wang, Jia Ruan, Jing Zhang, Chengjun Sun
TrAC Trends in Analytical Chemistry 2015 Volume 72() pp:10-26
Publication Date(Web):October 2015
DOI:10.1016/j.trac.2015.03.018
•Properties, toxicities and human exposure of phthalate esters.•Sample-preparation methods for analysis of phthalate esters.•Determination methods for phthalate esters in foods.•The impact of contamination on the analysis of phthalate esters.•Future trends in the analysis of phthalate esters in foods.Phthalate esters (PAEs) are widely used as plasticizers in food processing and packaging. Because of a growing international concern about the health effects of PAEs, the analysis of these compounds in various foods was one focus of research in recent years. This review provides an updated overview of recent advances in sample-preparation and determination methods for analysis of PAEs in foods. We discuss contamination problems, current challenges and future trends in the analysis of PAEs in foods.
Co-reporter:Juan Xie, Yongxin Li, Jing Zhang, Li Zeng, Dan Lu, Yapan Liu, Yi Yang and Chengjun Sun
Analytical Methods 2014 vol. 6(Issue 14) pp:5140-5146
Publication Date(Web):07 May 2014
DOI:10.1039/C4AY00509K
A simple, rapid, inexpensive and efficient method based on ultrasound-assisted dispersive liquid–liquid microextraction (UDLLME) after pre-column derivatization coupled with high performance capillary electrophoresis (HPCE) has been developed for determination of methylamine (MA), ethylamine (EA), dimethylamine (DMA) and diethylamine (DEA) in aquatic products. The aliphatic amines were derivatized with 9-fluorenyl methyl chloroformate (FMOC-Cl) in alkaline aqueous solution and then the derivatives were extracted by ultrasound-assisted liquid–liquid microextraction (UDLLME) with trichloromethane. The factors affecting the derivatization and extraction efficiencies were investigated in detail. The electrophoresis separation was performed in an uncoated fused-silica capillary (50 cm × 50 μm i.d.) with an effective length of 41 cm; 25 mmol L−1 sodium tetraborate containing 15 mmol L−1 sodium dodecyl sulfate (SDS) was used as running buffer; the applied voltage was 20 kV; the separation temperature was set to 25 °C; the UV detection wavelength was set at 265 nm. Four derivatized products of aliphatic amines could be completely separated within 7 min. The calibration curves were linear in the range of 0.5 to 12.5 mg L−1 for MA and DMA, and 1.0 to 25 mg L−1 for EA and DEA with the correlation coefficients ranging from 0.9984 to 0.9994. The limits of detection (LODs) and limits of quantification (LOQs) of the method were in the ranges of 0.028–0.16 mg kg−1 and 0.095–0.53 mg kg−1, respectively. The enrichment factors ranged from 42 to 68 for the four derivatives. The recoveries of the method were in the range of 72.8–97.0%, with the intraday relative standard deviations (RSDs) of the peak area in the range of 2.80–4.61%. This method has been successfully applied to the analysis of the four aliphatic amines in aquatic products.
Co-reporter:Fan YOU, Lan ZHU, Ling HE, Liang-Ji RAN, Yan JIN, Cheng-Jun SUN
Chinese Journal of Analytical Chemistry 2014 Volume 42(Issue 12) pp:1723-1728
Publication Date(Web):December 2014
DOI:10.1016/S1872-2040(14)60789-1
A high performance liquid chromatographic method for simultaneous determination of seven metabolites of polycyclic aromatic hydrocarbons (PAHs), including 1-hydroxynaphthalene, 2-hydroxynaphthalene, 2-hydroxyfluorene, 2-hydroxyphenanthrene, 4-hydroxyphenanthrene, 1-hydroxypyrene and 6-hydroxychrysene in human urine, was developed coupled with online solid phase extraction by use of a double ternary liquid chromatography system with a fluorescence detector. The target compounds were online concentrated on the Turboflow Cyclone solid phase extraction column, and then transferred by a six-way valve to the Hypersil Green PAH column for separation with acetonitrile and water as mobile phase at a flow rate of 1.0 mL min−1 and 35 °C. The analysis was completed in 20 min. Under the optimized chromatographic conditions, the method showed good linearity (r ≥ 0.999) in the range of 5−2000 ng L−1 or 50−20000 ng L−1 with the LODs of 0.5−15 ng L−1 and the recoveries of 80.7%−110.7%. The proposed method was successfully applied to the detection of metabolites of polycyclic aromatic hydrocarbons in urine from several smokers and non-smokers. The concentrations of 2-hydroxynaphthalene, 1-hydroxynaphthalene, 2-hydroxyfluorene, 2-hydroxyphenanthrene, 4-hydroxyphenanthrene and 6-hydroxychrysene in the smoker urine were much higher than those in non-smoker.A high performance liquid chromatographic method for simultaneous determination of seven metabolites of polycyclic aromatic hydrocarbons (PAHs), including 1-hydroxynaphthalene, 2-hydroxynaphthalene, 2-hydroxyfluorene, 2-hydroxyphenanthrene, 4-hydroxyphenanthrene, 1-hydroxypyrene and 6-hydroxychrysene in human urine, was developed coupled with online solid phase extraction by use of a double ternary liquid chromatography system with a fluorescence detector.
Co-reporter:Jinling Yang;Yongxin Li;Weilei Gong;Changqin Wang
Food Analytical Methods 2014 Volume 7( Issue 8) pp:1693-1702
Publication Date(Web):2014 September
DOI:10.1007/s12161-014-9807-9
A novel microextraction method termed matrix liquid-phase dispersion extraction (MLPDE) was presented in this paper. This method was adopted for the extraction of preservatives including methylparaben, ethylparaben, propylparaben, butylparaben, isopropylparaben, and isobutylparaben from different solid food samples. The extracted analytes were subjected to high-performance liquid chromatography (HPLC) analysis immediately. In this method, the food sample (0.50 g) was added previously to a syringe and then rinsed with hot water (45 °C). Subsequently, 2 ml extraction solvent (methanol) was added into the syringe. After ultrasonic extraction, food sample was dispersed into the methanol phase and a cloudy suspension came into being. Under the pressure of the plunger, methanol carrying the target analytes passed through 0.45 μm membrane and 10.0 μl of the eluent was injected into HPLC system. The important parameters including temperature, pH and volume of water, extraction solvent and its volume, sodium chloride addition, sample weight, and ultrasonic time as well as matrix effect were investigated. Under the optimum conditions, recoveries of the studied preservatives ranged from 93.7 % to 107.9 %. The intra-day and inter-day precisions (relative standard deviations) were less than 5.16 % and 5.32 %, respectively. The linear ranges of the method were 0.947–400 mg/kg with the correlation coefficients of 0.9988 to 0.9999. The limits of detection were 0.285 to 1.122 mg/kg. MLPDE is a very simple, rapid, cheap, and efficient method for extraction of nonpolar organic compounds from solid food samples. The proposed method has been successfully applied to the separation and quantification of six parabens in different solid food samples.
Co-reporter:
Analytical Methods (2009-Present) 2014 - vol. 6(Issue 14) pp:
Publication Date(Web):
DOI:10.1039/C4AY00509K
A simple, rapid, inexpensive and efficient method based on ultrasound-assisted dispersive liquid–liquid microextraction (UDLLME) after pre-column derivatization coupled with high performance capillary electrophoresis (HPCE) has been developed for determination of methylamine (MA), ethylamine (EA), dimethylamine (DMA) and diethylamine (DEA) in aquatic products. The aliphatic amines were derivatized with 9-fluorenyl methyl chloroformate (FMOC-Cl) in alkaline aqueous solution and then the derivatives were extracted by ultrasound-assisted liquid–liquid microextraction (UDLLME) with trichloromethane. The factors affecting the derivatization and extraction efficiencies were investigated in detail. The electrophoresis separation was performed in an uncoated fused-silica capillary (50 cm × 50 μm i.d.) with an effective length of 41 cm; 25 mmol L−1 sodium tetraborate containing 15 mmol L−1 sodium dodecyl sulfate (SDS) was used as running buffer; the applied voltage was 20 kV; the separation temperature was set to 25 °C; the UV detection wavelength was set at 265 nm. Four derivatized products of aliphatic amines could be completely separated within 7 min. The calibration curves were linear in the range of 0.5 to 12.5 mg L−1 for MA and DMA, and 1.0 to 25 mg L−1 for EA and DEA with the correlation coefficients ranging from 0.9984 to 0.9994. The limits of detection (LODs) and limits of quantification (LOQs) of the method were in the ranges of 0.028–0.16 mg kg−1 and 0.095–0.53 mg kg−1, respectively. The enrichment factors ranged from 42 to 68 for the four derivatives. The recoveries of the method were in the range of 72.8–97.0%, with the intraday relative standard deviations (RSDs) of the peak area in the range of 2.80–4.61%. This method has been successfully applied to the analysis of the four aliphatic amines in aquatic products.