Co-reporter:Claire M. Weekley, Isabell Kenkel, Rainer Lippert, Shengwei Wei, Dominik Lieb, Tiffanny Cranwell, Jason L. Wedding, Annika S. Zillmann, Robin Rohr, Milos R. Filipovic, Ivana Ivanović-Burmazović, and Hugh H. Harris
Inorganic Chemistry June 5, 2017 Volume 56(Issue 11) pp:6076-6076
Publication Date(Web):May 11, 2017
DOI:10.1021/acs.inorgchem.6b03073
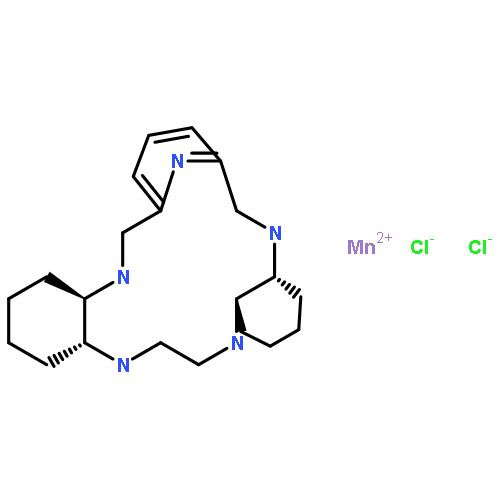
Manganese(II) pentaazamacrocyclic complexes (MnPAMs) can act as small-molecule mimics of manganese superoxide dismutase (MnSOD) with potential therapeutic application in conditions linked to oxidative stress. Previously, the in vitro mechanism of action has been determined, their activity has been demonstrated in cells, and some representatives of this class of MnSOD mimetics have entered clinical trials. However, MnPAM uptake, distribution, and metabolism in cells are largely unknown. Therefore, we have used X-ray fluorescence microscopy (XFM) and X-ray absorption spectroscopy (XAS) to study the cellular fate of a number of MnPAMs. We have also synthesized and characterized fluorescently labeled (pyrene and rhodamine) manganese(II) pyane [manganese(II) trans-2,13-dimethyl-3,6,9,12,18-pentaazabicyclo[12.3.1]octadeca-1(18),14,16-triene] derivatives and investigated their utility for cellular imaging of MnPAMs. Their SOD activity was determined via a direct stopped-flow technique. XFM experiments show that treatment with amine-based manganese(II) pyane type pentaazamacrocycles leads to a 10–100-fold increase in the overall cellular manganese levels compared to the physiological levels of manganese in control cells. In treated cells in general, manganese was distributed throughout the cell body, with a couple of notable exceptions. The lipophilicity of the MnPAMs, examined by partitioning in octanol–buffer system, was a good predictor of the relative cellular manganese levels. Analysis of the XAS data of treated cells revealed that some fraction of amine-based MnPAMs taken up by the cells remained intact, with the rest transformed into SOD-active manganese(II) phosphate. Higher phosphate binding constants, determined from the effect of the phosphate concentration on in vitro SOD activity, were associated with more extensive metabolism of the amine-based MnPAMs to manganese(II) phosphate. In contrast, the imine-based manganese(II) pydiene complex that is prone to hydrolysis was entirely decomposed after uptake and free manganese(II) was oxidized to a manganese(III) oxide type species, in cytosolic compartments, possibly mitochondria. Complex stability constants (determined for some of the MnPAMs) are less indicative of the cellular fate of the complexes than the corresponding phosphate binding constants.
Co-reporter:Maximilian Dürr, Johannes Klein, Axel Kahnt, Sabine Becker, Ralph Puchta, Biprajit Sarkar, and Ivana Ivanović-Burmazović
Inorganic Chemistry December 18, 2017 Volume 56(Issue 24) pp:14912-14912
Publication Date(Web):November 20, 2017
DOI:10.1021/acs.inorgchem.7b02192
A dinuclear ruthenium complex bridged by 2,3,5,6-pyrazinetetracarboxylic acid (μ-LH22–) was synthesized and characterized by X-ray crystallography, cyclic voltammetry under ambient and elevated pressures, electron paramagnetic resonance (EPR) and UV/vis-NIR (NIR = near-infrared) spectroelectrochemistry, pulse radiolysis, and computational methods. We probed for the first time in the field of mixed-valency the use of high-pressure electrochemical methods. The investigations were directed toward the influence of the protonation state of the bridging ligand on the electronic communication between the ruthenium ions, since such behavior is interesting in terms of modulating redox chemistry by pH. Starting from the [RuII(μ-LH22–)RuII]0 configuration, which shows an intense metal-to-ligand charge transfer absorption band at 600 nm, cyclic voltammetry revealed a pH-independent, reversible one-electron reduction and a protonation-state-dependent (proton coupled electron transfer, PCET) reversible oxidation. Deeper insight into the electrode reactions was provided by pressure-dependent cyclic voltammetry up to 150 MPa, providing insight into the conformational changes, the protonation state, and the environment of the molecule during the redox processes. Spectroelectrochemical investigations (EPR, UV/vis-NIR) of the respective redox reactions suggest a ligand-centered radical anion [RuII(μ-LH2•3–)RuII]− upon reduction (EPR Δg = 0.042) and an ambiguous, EPR-silent one-electron oxidized state. In both cases, the absence of the otherwise typical broad intervalence charge transfer bands in the NIR region for mixed-valent complexes support the formulation as radical anionic bridged compound. However, on the basis of high-pressure electrochemical data and density functional theory calculations the one-electron oxidized form could be assigned as a charge-delocalized [RuII.5(μ-LH22–)RuII.5]+ valence tautomer rather than [RuIII(μ-LH2•3–)RuIII]+. Deprotonation of the bridging ligand causes a severe shift of the redox potential for the metal-based oxidation toward lower potentials, yielding the charge-localized [RuIII(μ-LH3–)RuII]0 complex. This PCET process is accompanied by large intrinsic volume changes. All findings are supported by computational methods (geometry optimization, spin population analysis). For all redox processes, valence alternatives are discussed.
Co-reporter:Alison C. McQuilken; Hirotoshi Matsumura; Maximilian Dürr; Alex M. Confer; John P. Sheckelton; Maxime A. Siegler; Tyrel M. McQueen; Ivana Ivanović-Burmazović; Pierre Moënne-Loccoz;David P. Goldberg
Journal of the American Chemical Society 2016 Volume 138(Issue 9) pp:3107-3117
Publication Date(Web):February 26, 2016
DOI:10.1021/jacs.5b12741
The nonheme iron complex, [Fe(NO)(N3PyS)]BF4, is a rare example of an {FeNO}7 species that exhibits spin-crossover behavior. The comparison of X-ray crystallographic studies at low and high temperatures and variable-temperature magnetic susceptibility measurements show that a low-spin S = 1/2 ground state is populated at 0–150 K, while both low-spin S = 1/2 and high-spin S = 3/2 states are populated at T > 150 K. These results explain the observation of two N–O vibrational modes at 1737 and 1649 cm–1 in CD3CN for [Fe(NO)(N3PyS)]BF4 at room temperature. This {FeNO}7 complex reacts with dioxygen upon photoirradiation with visible light in acetonitrile to generate a thiolate-ligated, nonheme iron(III)-nitro complex, [FeIII(NO2)(N3PyS)]+, which was characterized by EPR, FTIR, UV–vis, and CSI-MS. Isotope labeling studies, coupled with FTIR and CSI-MS, show that one O atom from O2 is incorporated in the FeIII–NO2 product. The O2 reactivity of [Fe(NO)(N3PyS)]BF4 in methanol is dramatically different from CH3CN, leading exclusively to sulfur-based oxidation, as opposed to NO· oxidation. A mechanism is proposed for the NO· oxidation reaction that involves formation of both FeIII-superoxo and FeIII-peroxynitrite intermediates and takes into account the experimental observations. The stability of the FeIII-nitrite complex is limited, and decay of [FeIII(NO2)(N3PyS)]+ leads to {FeNO}7 species and sulfur oxygenated products. This work demonstrates that a single mononuclear, thiolate-ligated nonheme {FeNO}7 complex can exhibit reactivity related to both nitric oxide dioxygenase (NOD) and nitrite reductase (NiR) activity. The presence of the thiolate donor is critical to both pathways, and mechanistic insights into these biologically relevant processes are presented.
Co-reporter:Shabnam Hematian; Isabell Kenkel; Tatyana E. Shubina; Maximilian Dürr; Jeffrey J. Liu; Maxime A. Siegler; Ivana Ivanovic-Burmazovic;Kenneth D. Karlin
Journal of the American Chemical Society 2015 Volume 137(Issue 20) pp:6602-6615
Publication Date(Web):May 14, 2015
DOI:10.1021/jacs.5b02174
While nitric oxide (NO, nitrogen monoxide) is a critically important signaling agent, its cellular concentrations must be tightly controlled, generally through its oxidative conversion to nitrite (NO2–) where it is held in reserve to be reconverted as needed. In part, this reaction is mediated by the binuclear heme a3/CuB active site of cytochrome c oxidase. In this report, the oxidation of NO(g) to nitrite is shown to occur efficiently in new synthetic μ-oxo heme-FeIII–O–CuII(L) constructs (L being a tridentate or tetradentate pyridyl/alkylamino ligand), and spectroscopic and kinetic investigations provide detailed mechanistic insights. Two new X-ray structures of μ-oxo complexes have been determined and compared to literature analogs. All μ-oxo complexes react with 2 mol equiv NO(g) to give 1:1 mixtures of discrete [(L)CuII(NO2–)]+ plus ferrous heme-nitrosyl compounds; when the first NO(g) equiv reduces the heme center and itself is oxidized to nitrite, the second equiv of NO(g) traps the ferrous heme thus formed. For one μ-oxo heme-FeIII–O–CuII(L) compound, the reaction with NO(g) reveals an intermediate species (“intermediate”), formally a bis-NO adduct, [(NO)(porphyrinate)FeII–(NO2–)–CuII(L)]+ (λmax = 433 nm), confirmed by cryo-spray ionization mass spectrometry and EPR spectroscopy, along with the observation that cooling a 1:1 mixture of [(L)CuII(NO2–)]+ and heme-FeII(NO) to −125 °C leads to association and generation of the key 433 nm UV–vis feature. Kinetic-thermodynamic parameters obtained from low-temperature stopped-flow measurements are in excellent agreement with DFT calculations carried out which describe the sequential addition of NO(g) to the μ-oxo complex.
Co-reporter:Anne Dees; Norbert Jux; Oliver Tröppner; Katharina Dürr; Rainer Lippert; Martin Schmid; Bernd Küstner; Sebastian Schlücker; Hans-Peter Steinrück; J. Michael Gottfried;Ivana Ivanović-Burmazović
Inorganic Chemistry 2015 Volume 54(Issue 14) pp:6862-6872
Publication Date(Web):July 9, 2015
DOI:10.1021/acs.inorgchem.5b00770
The redox reaction of superoxide (KO2) with highly charged iron porphyrins (Fe(P4+), Fe(P8+), and Fe(P8−)) has been investigated in the ionic liquids (IL) [EMIM][Tf2N] (1-ethyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide) and [EMIM][B(CN)4] (1-ethyl-3-methylimidazolium tetracyanoborate) by using time-resolved UV/vis stopped-flow, electrochemistry, cryospray mass spectrometry, EPR, and XPS measurements. Stable KO2 solutions in [EMIM][Tf2N] can be prepared up to a 15 mM concentration and are characterized by a signal in EPR spectrum at g = 2.0039 and by the 1215 cm–1 stretching vibration in the resonance Raman spectrum. While the negatively charged iron porphyrin Fe(P8−) does not react with superoxide in IL, Fe(P4+) and Fe(P8+) do react in a two-step process (first a reduction of the Fe(III) to the Fe(II) form, followed by the binding of superoxide to Fe(II)). In the reaction with KO2, Fe(P4+) and Fe(P8+) show similar rate constants (e.g., in the case of Fe(P4+): k1 = 18.6 ± 0.5 M–1 s–1 for the first reaction step, and k2 = 2.8 ± 0.1 M–1 s–1 for the second reaction step). Notably, these rate constants are four to five orders of magnitude lower in [EMIM][Tf2N] than in conventional solvents such as DMSO. The influence of the ionic liquid is also apparent during electrochemical experiments, where the redox potentials for the corresponding Fe(III)/Fe(II) couples are much more negative in [EMIM][Tf2N] than in DMSO. This modified redox and kinetic behavior of the positively charged iron porphyrins results from their interactions with the anions of the ionic liquid, while the nucleophilicity of the superoxide is reduced by its interactions with the cations of the ionic liquid. A negligible vapor pressure of [EMIM][B(CN)4] and a sufficient enrichment of Fe(P8+) in a close proximity to the surface enabled XPS measurements as a case study for monitoring direct changes in the electronic structure of the metal centers during redox processes in solution and at liquid/solid interfaces.
Co-reporter:Rudolf Wedmann; Achim Zahl; Tatyana E. Shubina; Maximilian Dürr; Frank W. Heinemann; Bernhard Eberhard Christian Bugenhagen; Peter Burger; Ivana Ivanovic-Burmazovic;Milos R. Filipovic
Inorganic Chemistry 2015 Volume 54(Issue 19) pp:9367-9380
Publication Date(Web):June 26, 2015
DOI:10.1021/acs.inorgchem.5b00831
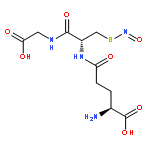
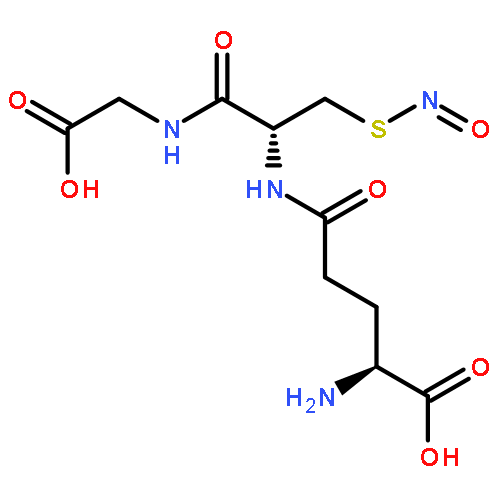
Hydrogen sulfide (H2S) and nitric oxide (NO) are important signaling molecules that regulate several physiological functions. Understanding the chemistry behind their interplay is important for explaining these functions. The reaction of H2S with S-nitrosothiols to form the smallest S-nitrosothiol, thionitrous acid (HSNO), is one example of physiologically relevant cross-talk between H2S and nitrogen species. Perthionitrite (SSNO–) has recently been considered as an important biological source of NO that is far more stable and longer living than HSNO. In order to experimentally address this issue here, we prepared SSNO– by two different approaches, which lead to two distinct species: SSNO– and dithionitric acid [HON(S)S/HSN(O)S]. (H)S2NO species and their reactivity were studied by 15N NMR, IR, electron paramagnetic resonance and high-resolution electrospray ionization time-of-flight mass spectrometry, as well as by X-ray structure analysis and cyclic voltammetry. The obtained results pointed toward the inherent instability of SSNO– in water solutions. SSNO– decomposed readily in the presence of light, water, or acid, with concomitant formation of elemental sulfur and HNO. Furthermore, SSNO− reacted with H2S to generate HSNO. Computational studies on (H)SSNO provided additional explanations for its instability. Thus, on the basis of our data, it seems to be less probable that SSNO– can serve as a signaling molecule and biological source of NO. SSNO– salts could, however, be used as fast generators of HNO in water solutions.
Co-reporter:O. Troeppner, D. Huang, R. H. Holm and I. Ivanović-Burmazović
Dalton Transactions 2014 vol. 43(Issue 14) pp:5274-5279
Publication Date(Web):26 Feb 2014
DOI:10.1039/C3DT53004C
The previously reported carbon dioxide fixation reaction by the planar terminal hydroxide complex [Ni(pyN2Me2)(OH)]1− in DMF has been further characterized by determination of the equilibrium constants K298eq = 2.4 ± 0.2 × 105 M−1 and K223eq = 1.3 ± 0.1 × 107 M−1, as well as the volume of activation for the CO2 binding (ΔV≠223on = −21 ± 3 cm3 mol−1) and back decarboxylation (ΔV≠223off = −13 ± 1 cm3 mol−1) by high-pressure kinetics. The data are consistent with an earlier DFT computation, including the probable nature of the transition state, and support designating the reaction as one of the most completely investigated carbon dioxide fixation reactions of any type.
Co-reporter:Dipl.-Chem. Oliver Troeppner;Dr. Rainer Lippert;Dr. Tatyana E. Shubina;Dr. Achim Zahl;Dr. Norbert Jux;Dr. Ivana Ivanovi&x107;-Burmazovi&x107;
Angewandte Chemie 2014 Volume 126( Issue 43) pp:11636-11641
Publication Date(Web):
DOI:10.1002/ange.201406954
Abstract
By design of a heme model complex with a binding pocket of appropriate size and flexibility, and by elucidating its kinetics and thermodynamics under elevated pressures, some of the pressure effects are demonstrated relevant for operation of heme-proteins under deep-sea conditions. Opposite from classical paradigms of the spin-crossover and reaction kinetics, a pressure increase can cause deceleration of the small-molecule binding to the vacant coordination site of the heme-center in a confined space and stabilize a high-spin state of its Fe center. This reverse high-pressure behavior can be achieved only if the volume changes related to the conformational transformation of the cavity can offset the volume changes caused by the substrate binding. It is speculated that based on these criteria nature could make a selection of structures of heme pockets that assist in reducing metabolic activity and enzymatic side reactions under extreme pressure conditions.
Co-reporter:Dipl.-Chem. Oliver Troeppner;Dr. Rainer Lippert;Dr. Tatyana E. Shubina;Dr. Achim Zahl;Dr. Norbert Jux;Dr. Ivana Ivanovi&x107;-Burmazovi&x107;
Angewandte Chemie 2014 Volume 126( Issue 43) pp:
Publication Date(Web):
DOI:10.1002/ange.201484361
Co-reporter:Dipl.-Chem. Oliver Troeppner;Dr. Rainer Lippert;Dr. Tatyana E. Shubina;Dr. Achim Zahl;Dr. Norbert Jux;Dr. Ivana Ivanovi&x107;-Burmazovi&x107;
Angewandte Chemie International Edition 2014 Volume 53( Issue 43) pp:11452-11457
Publication Date(Web):
DOI:10.1002/anie.201406954
Abstract
By design of a heme model complex with a binding pocket of appropriate size and flexibility, and by elucidating its kinetics and thermodynamics under elevated pressures, some of the pressure effects are demonstrated relevant for operation of heme-proteins under deep-sea conditions. Opposite from classical paradigms of the spin-crossover and reaction kinetics, a pressure increase can cause deceleration of the small-molecule binding to the vacant coordination site of the heme-center in a confined space and stabilize a high-spin state of its Fe center. This reverse high-pressure behavior can be achieved only if the volume changes related to the conformational transformation of the cavity can offset the volume changes caused by the substrate binding. It is speculated that based on these criteria nature could make a selection of structures of heme pockets that assist in reducing metabolic activity and enzymatic side reactions under extreme pressure conditions.
Co-reporter:Dipl.-Chem. Oliver Troeppner;Dr. Rainer Lippert;Dr. Tatyana E. Shubina;Dr. Achim Zahl;Dr. Norbert Jux;Dr. Ivana Ivanovi&x107;-Burmazovi&x107;
Angewandte Chemie International Edition 2014 Volume 53( Issue 43) pp:
Publication Date(Web):
DOI:10.1002/anie.201484361
Co-reporter:Jan Lj. Miljkovic;Isabell Kenkel;Dr. Ivana Ivanovi&x107;-Burmazovi&x107;;Dr. Milos R. Filipovic
Angewandte Chemie International Edition 2013 Volume 52( Issue 46) pp:12061-12064
Publication Date(Web):
DOI:10.1002/anie.201305669
Co-reporter:Jan Lj. Miljkovic;Isabell Kenkel;Dr. Ivana Ivanovi&x107;-Burmazovi&x107;;Dr. Milos R. Filipovic
Angewandte Chemie 2013 Volume 125( Issue 46) pp:12283-12286
Publication Date(Web):
DOI:10.1002/ange.201305669
Co-reporter:O. Troeppner, D. Huang, R. H. Holm and I. Ivanović-Burmazović
Dalton Transactions 2014 - vol. 43(Issue 14) pp:NaN5279-5279
Publication Date(Web):2014/02/26
DOI:10.1039/C3DT53004C
The previously reported carbon dioxide fixation reaction by the planar terminal hydroxide complex [Ni(pyN2Me2)(OH)]1− in DMF has been further characterized by determination of the equilibrium constants K298eq = 2.4 ± 0.2 × 105 M−1 and K223eq = 1.3 ± 0.1 × 107 M−1, as well as the volume of activation for the CO2 binding (ΔV≠223on = −21 ± 3 cm3 mol−1) and back decarboxylation (ΔV≠223off = −13 ± 1 cm3 mol−1) by high-pressure kinetics. The data are consistent with an earlier DFT computation, including the probable nature of the transition state, and support designating the reaction as one of the most completely investigated carbon dioxide fixation reactions of any type.



![Thiourea, N-[(1S,2S)-2-aminocyclohexyl]-N'-[(1R)-1-phenylethyl]-](/data/chemimg/1372000/874662-43-8.png)
![Thiourea, N-[(1S,2S)-2-aminocyclohexyl]-N'-[(1R)-1-phenylethyl]-](/data/chemimg/1372000/874662-43-8_b.png)








![21H,23H-Porphine, 5,10,15,20-tetrakis[4-(1,1-dimethylethyl)phenyl]-](/data/chemimg/133300/110452-48-7.png)
![21H,23H-Porphine, 5,10,15,20-tetrakis[4-(1,1-dimethylethyl)phenyl]-](/data/chemimg/133300/110452-48-7_b.png)
![Manganese(5 ), [[4,4',4'',4'''-(21H,23H-porphine-5,10,15,20-tetrayl-κN21,κN22,κN23,κN24)tetrakis[1-methylpyridiniumato]](2-)]-, (SP-4-1)-](/data/chemimg/1563200/70649-54-6.png)
![Manganese(5 ), [[4,4',4'',4'''-(21H,23H-porphine-5,10,15,20-tetrayl-κN21,κN22,κN23,κN24)tetrakis[1-methylpyridiniumato]](2-)]-, (SP-4-1)-](/data/chemimg/1563200/70649-54-6_b.png)
![Benzeneacetic acid,4-hydroxy-3-methoxy-,[(2S,3aR,3bS,6aR,9aR,9bR,10R,11aR)-3a,3b,6,6a,9a,10,11,11a-octahydro-6a-hydroxy-8,10-dimethyl-11a-(1-methylethenyl)-7-oxo-2-(phenylmethyl)-7H-2,9b-epoxyazuleno[5,4-e]-1,3-benzodioxol-5-yl]methylester](http://img.cochemist.com/ccimg/57500/57444-62-9.png)
![Benzeneacetic acid,4-hydroxy-3-methoxy-,[(2S,3aR,3bS,6aR,9aR,9bR,10R,11aR)-3a,3b,6,6a,9a,10,11,11a-octahydro-6a-hydroxy-8,10-dimethyl-11a-(1-methylethenyl)-7-oxo-2-(phenylmethyl)-7H-2,9b-epoxyazuleno[5,4-e]-1,3-benzodioxol-5-yl]methylester](http://img.cochemist.com/ccimg/57500/57444-62-9_b.png)
![dihydrogen [[N,N'-ethylenebis[N-(carboxymethyl)glycinato]](4-)-N,N',O,O',ON,ON']manganate(2-)](http://img.cochemist.com/ccimg/55500/55448-20-9.png)
![dihydrogen [[N,N'-ethylenebis[N-(carboxymethyl)glycinato]](4-)-N,N',O,O',ON,ON']manganate(2-)](http://img.cochemist.com/ccimg/55500/55448-20-9_b.png)





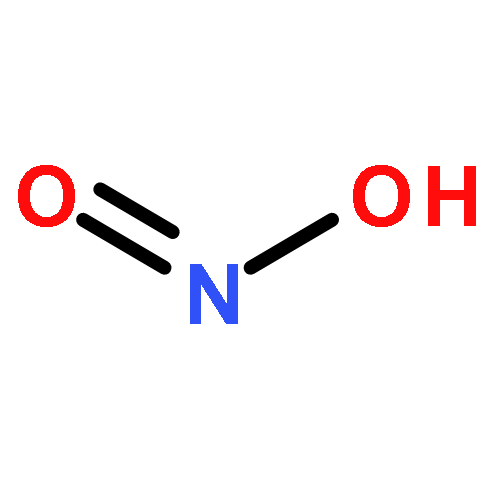
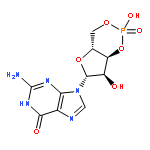
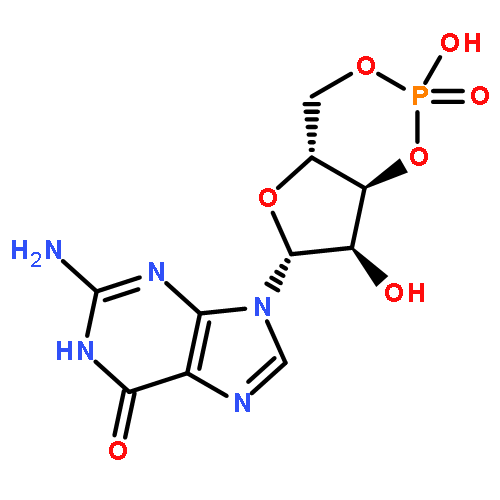
![6H-Benzo[cd]pyren-6-one](http://img.cochemist.com/ccimg/3100/3074-00-8.png)
![6H-Benzo[cd]pyren-6-one](http://img.cochemist.com/ccimg/3100/3074-00-8_b.png)
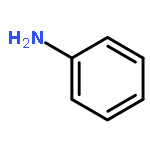






![Ferrate(2-), [7,12-diethenyl-3,8,13,17-tetramethyl-21H,23H-porphine-2,18-dipropanoato(4-)-κN21,κN22,κN23,κN24]-, hydrogen (1:2), (SP-4-2)-](/data/chemimg/522800/14875-96-8.png)
![Ferrate(2-), [7,12-diethenyl-3,8,13,17-tetramethyl-21H,23H-porphine-2,18-dipropanoato(4-)-κN21,κN22,κN23,κN24]-, hydrogen (1:2), (SP-4-2)-](/data/chemimg/522800/14875-96-8_b.png)