Co-reporter:David W. Christianson
Chemical Reviews September 13, 2017 Volume 117(Issue 17) pp:11570-11570
Publication Date(Web):August 25, 2017
DOI:10.1021/acs.chemrev.7b00287
The year 2017 marks the twentieth anniversary of terpenoid cyclase structural biology: a trio of terpenoid cyclase structures reported together in 1997 were the first to set the foundation for understanding the enzymes largely responsible for the exquisite chemodiversity of more than 80000 terpenoid natural products. Terpenoid cyclases catalyze the most complex chemical reactions in biology, in that more than half of the substrate carbon atoms undergo changes in bonding and hybridization during a single enzyme-catalyzed cyclization reaction. The past two decades have witnessed structural, functional, and computational studies illuminating the modes of substrate activation that initiate the cyclization cascade, the management and manipulation of high-energy carbocation intermediates that propagate the cyclization cascade, and the chemical strategies that terminate the cyclization cascade. The role of the terpenoid cyclase as a template for catalysis is paramount to its function, and protein engineering can be used to reprogram the cyclization cascade to generate alternative and commercially important products. Here, I review key advances in terpenoid cyclase structural and chemical biology, focusing mainly on terpenoid cyclases and related prenyltransferases for which X-ray crystal structures have informed and advanced our understanding of enzyme structure and function.
Co-reporter:Patrick N. Blank, Golda H. Barrow, Wayne K. W. Chou, Lian Duan, David E. Cane, and David W. Christianson
Biochemistry October 31, 2017 Volume 56(Issue 43) pp:5798-5798
Publication Date(Web):October 2, 2017
DOI:10.1021/acs.biochem.7b00895
The sesquiterpene cyclase epi-isozizaene synthase (EIZS) catalyzes the cyclization of farnesyl diphosphate to form the tricyclic hydrocarbon precursor of the antibiotic albaflavenone. The hydrophobic active site pocket of EIZS serves as a template as it binds and chaperones the flexible substrate and carbocation intermediates through the conformations required for a multistep reaction sequence. We previously demonstrated that the substitution of hydrophobic residues with other hydrophobic residues remolds the template and expands product chemodiversity [Li, R., Chou, W. K. W., Himmelberger, J. A., Litwin, K. M., Harris, G. G., Cane, D. E., and Christianson, D. W. (2014) Biochemistry 53, 1155–1168]. Here, we show that the substitution of hydrophobic residues—specifically, Y69, F95, F96, and W203—with polar side chains also yields functional enzyme catalysts that expand product chemodiversity. Fourteen new EIZS mutants are reported that generate product arrays in which eight new sesquiterpene products have been identified. Of note, some mutants generate acyclic and cyclic hydroxylated products, suggesting that the introduction of polarity in the hydrophobic pocket facilitates the binding of water capable of quenching carbocation intermediates. Furthermore, the substitution of polar residues for F96 yields high-fidelity sesquisabinene synthases. Crystal structures of selected mutants reveal that residues defining the three-dimensional contour of the hydrophobic pocket can be substituted without triggering significant structural changes elsewhere in the active site. Thus, more radical nonpolar–polar amino acid substitutions should be considered when terpenoid cyclase active sites are remolded by mutagenesis with the goal of exploring and expanding product chemodiversity.
Co-reporter:Mengbin Chen, Wayne K. W. Chou, Tomonobu Toyomasu, David E. Cane, and David W. Christianson
ACS Chemical Biology 2016 Volume 11(Issue 4) pp:889
Publication Date(Web):January 6, 2016
DOI:10.1021/acschembio.5b00960
Fusicoccin A is a diterpene glucoside phytotoxin generated by the fungal pathogen Phomopsis amygdali that causes the plant disease constriction canker, first discovered in New Jersey peach orchards in the 1930s. Fusicoccin A is also an emerging new lead in cancer chemotherapy. The hydrocarbon precursor of fusicoccin A is the tricyclic diterpene fusicoccadiene, which is generated by a bifunctional terpenoid synthase. Here, we report X-ray crystal structures of the individual catalytic domains of fusicoccadiene synthase: the C-terminal domain is a chain elongation enzyme that generates geranylgeranyl diphosphate, and the N-terminal domain catalyzes the cyclization of geranylgeranyl diphosphate to form fusicoccadiene. Crystal structures of each domain complexed with bisphosphonate substrate analogues suggest that three metal ions and three positively charged amino acid side chains trigger substrate ionization in each active site. While in vitro incubations reveal that the cyclase domain can utilize farnesyl diphosphate and geranyl diphosphate as surrogate substrates, these shorter isoprenoid diphosphates are mainly converted into acyclic alcohol or hydrocarbon products. Gel filtration chromatography and analytical ultracentrifugation experiments indicate that full-length fusicoccadiene synthase adopts hexameric quaternary structure, and small-angle X-ray scattering data yield a well-defined molecular envelope illustrating a plausible model for hexamer assembly.
Co-reporter:Sister M. Lucy Gantt FSGM, Christophe Decroos, Matthew S. Lee, Laura E. Gullett, Christine M. Bowman, David W. Christianson, and Carol A. Fierke
Biochemistry 2016 Volume 55(Issue 5) pp:820-832
Publication Date(Web):January 25, 2016
DOI:10.1021/acs.biochem.5b01327
Histone deacetylases (HDACs) regulate cellular processes such as differentiation and apoptosis and are targeted by anticancer therapeutics in development and in the clinic. HDAC8 is a metal-dependent class I HDAC and is proposed to use a general acid–base catalytic pair in the mechanism of amide bond hydrolysis. Here, we report site-directed mutagenesis and enzymological measurements to elucidate the catalytic mechanism of HDAC8. Specifically, we focus on the catalytic function of Y306 and the histidine-aspartate dyads H142-D176 and H143-D183. Additionally, we report X-ray crystal structures of four representative HDAC8 mutants: D176N, D176N/Y306F, D176A/Y306F, and H142A/Y306F. These structures provide a useful framework for understanding enzymological measurements. The pH dependence of kcat/KM for wild-type Co(II)-HDAC8 is bell-shaped with two pKa values of 7.4 and 10.0. The upper pKa reflects the ionization of the metal-bound water molecule and shifts to 9.1 in Zn(II)-HDAC8. The H142A mutant has activity 230-fold lower than that of wild-type HDAC8, but the pKa1 value is not altered. Y306F HDAC8 is 150-fold less active than the wild-type enzyme; crystal structures show that Y306 hydrogen bonds with the zinc-bound substrate carbonyl, poised for transition state stabilization. The H143A and H142A/H143A mutants exhibit activity that is >80000-fold lower than that of wild-type HDAC8; the buried D176N and D176A mutants have significant catalytic effects, with more subtle effects caused by D183N and D183A. These enzymological and structural studies strongly suggest that H143 functions as a single general base–general acid catalyst, while H142 remains positively charged and serves as an electrostatic catalyst for transition state stabilization.
Co-reporter:Mengbin Chen, Wayne K. W. Chou, Naeemah Al-Lami, Juan A. Faraldos, Rudolf K. Allemann, David E. Cane, and David W. Christianson
Biochemistry 2016 Volume 55(Issue 20) pp:2864-2874
Publication Date(Web):May 12, 2016
DOI:10.1021/acs.biochem.6b00343
Aristolochene synthase (ATAS) is a high-fidelity terpenoid cyclase that converts farnesyl diphosphate exclusively into the bicyclic hydrocarbon aristolochene. Previously determined crystal structures of ATAS complexes revealed trapped active site water molecules that could potentially interact with catalytic intermediates: water “w” hydrogen bonds with S303 and N299, water molecules “w1” and “w2” hydrogen bond with Q151, and a fourth water molecule coordinates to the Mg2+C ion. There is no obvious role for water in the ATAS mechanism because the enzyme exclusively generates a hydrocarbon product. Thus, these water molecules are tightly controlled so that they cannot react with carbocation intermediates. Steady-state kinetics and product distribution analyses of eight ATAS mutants designed to perturb interactions with active site water molecules (S303A, S303H, S303D, N299A, N299L, N299A/S303A, Q151H, and Q151E) indicate relatively modest effects on catalysis but significant effects on sesquiterpene product distributions. X-ray crystal structures of S303A, N299A, N299A/S303A, and Q151H mutants reveal minimal perturbation of active site solvent structure. Seven of the eight mutants generate farnesol and nerolidol, possibly resulting from addition of the Mg2+C-bound water molecule to the initially formed farnesyl cation, but no products are generated that would suggest enhanced reactivity of other active site water molecules. However, intermediate germacrene A tends to accumulate in these mutants. Thus, apart from the possible reactivity of Mg2+C-bound water, active site water molecules in ATAS are not directly involved in the chemistry of catalysis but instead contribute to the template that governs the conformation of the flexible substrate and carbocation intermediates.
Co-reporter:Nicholas J. Porter, Nicolas H. Christianson, Christophe Decroos, and David W. Christianson
Biochemistry 2016 Volume 55(Issue 48) pp:
Publication Date(Web):November 11, 2016
DOI:10.1021/acs.biochem.6b01014
Histone deacetylase 8 (HDAC8) catalyzes the hydrolysis of acetyl-l-lysine to yield products l-lysine and acetate through a mechanism in which a nucleophilic water molecule is activated by a histidine general base and a catalytic metal ion (Zn2+ or Fe2+). Acetyl-l-lysine also requires activation by metal coordination and a hydrogen bond with catalytic tyrosine Y306, which also functions in transition state stabilization. Interestingly, Y306 is located in the conserved glycine-rich loop G302GGGY. The potential flexibility afforded by the tetraglycine segment may facilitate induced-fit conformational changes in Y306 between “in” and “out” positions, as observed in related deacetylases. To probe the catalytic importance of the glycine-rich loop in HDAC8, we rigidified this loop by preparing the G302A, G303A, G304A, and G305A mutants and measured their steady state kinetics and determined their X-ray crystal structures. Substantial losses of catalytic efficiency are observed (10–500-fold based on kcat/KM), particularly for G304A HDAC8 and G305A HDAC8. These mutants also exhibit the greatest structural changes for catalytic tyrosine Y306 (1.3–1.7 Å shifts of the phenolic hydroxyl group). Molecular dynamics simulations further indicate that G304 and G305 undergo pronounced structural changes as residue 306 undergoes a transition between “in” and “out” conformations. Thus, the G304A and G305A substitutions likely compromise the position and conformational changes of Y306 required for substrate activation and transition state stabilization. The G302A and G303A substitutions have less severe catalytic consequences, and these substitutions may influence an internal channel through which product acetate is believed to exit.
Co-reporter:Yang Hai
Acta Crystallographica Section F 2016 Volume 72( Issue 4) pp:300-306
Publication Date(Web):
DOI:10.1107/S2053230X16003630
Leishmania arginase is a potential drug target for the treatment of leishmaniasis because this binuclear manganese metalloenzyme initiates de novo polyamine biosynthesis by catalyzing the hydrolysis of l-arginine to generate l-ornithine and urea. The product l-ornithine subsequently undergoes decarboxylation to yield putrescine, which in turn is utilized for spermidine biosynthesis. Polyamines such as spermidine are essential for the growth and survival of the parasite, so inhibition of enzymes in the polyamine-biosynthetic pathway comprises an effective strategy for treating parasitic infections. To this end, two X-ray crystal structures of L. mexicana arginase complexed with α,α-disubstituted boronic amino-acid inhibitors based on the molecular scaffold of 2-(S)-amino-6-boronohexanoic acid are now reported. Structural comparisons with human and parasitic arginase complexes reveal interesting differences in the binding modes of the additional α-substituents, i.e. the d side chains, of these inhibitors. Subtle differences in the three-dimensional contours of the outer active-site rims among arginases from different species lead to different conformations of the d side chains and thus different inhibitor-affinity trends. The structures suggest that it is possible to maintain affinity while fine-tuning intermolecular interactions of the d side chain of α,α-disubstituted boronic amino-acid inhibitors in the search for isozyme-specific and species-specific arginase inhibitors.
Co-reporter:Travis A Pemberton and David W Christianson
The Journal of Antibiotics 2016 69(7) pp:486-493
Publication Date(Web):April 13, 2016
DOI:10.1038/ja.2016.39
Terpenoid cyclases catalyze the most complex reactions in biology, in that more than half of the substrate carbon atoms often undergo changes in bonding during the course of a multistep cyclization cascade that proceeds through multiple carbocation intermediates. Many cyclization mechanisms require stereospecific deprotonation and reprotonation steps, and most cyclization cascades are terminated by deprotonation to yield an olefin product. The first bacterial terpenoid cyclase to yield a crystal structure was pentalenene synthase from Streptomyces exfoliatus UC5319. This cyclase generates the hydrocarbon precursor of the pentalenolactone family of antibiotics. The structures of pentalenene synthase and other terpenoid cyclases reveal predominantly nonpolar active sites typically lacking amino acid side chains capable of serving general base-general acid functions. What chemical species, then, enables the Brønsted acid–base chemistry required in the catalytic mechanisms of these enzymes? The most likely candidate for such general base-general acid chemistry is the co-product inorganic pyrophosphate. Here, we briefly review biological and nonbiological systems in which phosphate and its derivatives serve general base and general acid functions in catalysis. These examples highlight the fact that the Brønsted acid–base activities of phosphate derivatives are comparable to the Brønsted acid–base activities of amino acid side chains.
Co-reporter:Christophe Decroos, Dane J. Clausen, Brandon E. Haines, Olaf Wiest, Robert M. Williams, and David W. Christianson
Biochemistry 2015 Volume 54(Issue 12) pp:2126-2135
Publication Date(Web):March 20, 2015
DOI:10.1021/acs.biochem.5b00010
The macrocyclic depsipeptide Largazole is a potent inhibitor of metal-dependent histone deacetylases (HDACs), some of which are drug targets for cancer chemotherapy. Indeed, Largazole partially resembles Romidepsin (FK228), a macrocyclic depsipeptide already approved for clinical use. Each inhibitor contains a pendant side chain thiol that coordinates to the active site Zn2+ ion, as observed in the X-ray crystal structure of the HDAC8–Largazole complex [Cole, K. E., Dowling, D. P., Boone, M. A., Phillips, A. J., and Christianson, D. W. (2011) J. Am. Chem. Soc. 133, 12474]. Here, we report the X-ray crystal structures of HDAC8 complexed with three synthetic analogues of Largazole in which the depsipeptide ester is replaced with a rigid amide linkage. In two of these analogues, a six-membered pyridine ring is also substituted (with two different orientations) for the five-membered thiazole ring in the macrocycle skeleton. The side chain thiol group of each analogue coordinates to the active site Zn2+ ion with nearly ideal geometry, thereby preserving the hallmark structural feature of inhibition by Largazole. Surprisingly, in comparison with the binding of Largazole, these analogues trigger alternative conformational changes in loops L1 and L2 flanking the active site. However, despite these structural differences, inhibitory potency is generally comparable to, or just moderately less than, the inhibitory potency of Largazole. Thus, this study reveals important new structure–affinity relationships for the binding of macrocyclic inhibitors to HDAC8.
Co-reporter:Christophe Decroos and David W. Christianson
Biochemistry 2015 Volume 54(Issue 30) pp:4692-4703
Publication Date(Web):July 22, 2015
DOI:10.1021/acs.biochem.5b00536
Polyamines are essential aliphatic polycations that bind to nucleic acids and accordingly are involved in a variety of cellular processes. Polyamine function can be regulated by acetylation and deacetylation, just as histone function can be regulated by lysine acetylation and deacetylation. Acetylpolyamine amidohydrolase (APAH) from Mycoplana ramosa is a zinc-dependent polyamine deacetylase that shares approximately 20% amino acid sequence identity with human histone deacetylases. We now report the X-ray crystal structures of APAH–inhibitor complexes in a new and superior crystal form that diffracts to very high resolution (1.1–1.4 Å). Inhibitors include previously synthesized analogues of N8-acetylspermidine bearing trifluoromethylketone, thiol, and hydroxamate zinc-binding groups [Decroos, C., Bowman, C. M., and Christianson, D. W. (2013) Bioorg. Med. Chem. 21, 4530], and newly synthesized hydroxamate analogues of shorter, monoacetylated diamines, the most potent of which is the hydroxamate analogue of N-acetylcadaverine (IC50 = 68 nM). The high-resolution crystal structures of APAH–inhibitor complexes provide key inferences about the inhibition and catalytic mechanism of zinc-dependent deacetylases. For example, the trifluoromethylketone analogue of N8-acetylspermidine binds as a tetrahedral gem-diol that mimics the tetrahedral intermediate and its flanking transition states in catalysis. Surprisingly, this compound is also a potent inhibitor of human histone deacetylase 8 with an IC50 of 260 nM. Crystal structures of APAH–inhibitor complexes are determined at the highest resolution of any currently existing zinc deacetylase structure and thus represent the most accurate reference points for understanding structure–mechanism and structure–inhibition relationships in this critically important enzyme family.
Co-reporter:Christophe Decroos, Nicolas H. Christianson, Laura E. Gullett, Christine M. Bowman, Karen E. Christianson, Matthew A. Deardorff, and David W. Christianson
Biochemistry 2015 Volume 54(Issue 42) pp:6501-6513
Publication Date(Web):October 14, 2015
DOI:10.1021/acs.biochem.5b00881
Cornelia de Lange Syndrome (CdLS) spectrum disorders are characterized by multiple organ system congenital anomalies that result from mutations in genes encoding core cohesin proteins SMC1A, SMC3, and RAD21, or proteins that regulate cohesin function such as NIPBL and HDAC8. HDAC8 is the Zn2+-dependent SMC3 deacetylase required for cohesin recycling during the cell cycle, and 17 different HDAC8 mutants have been identified to date in children diagnosed with CdLS. As part of our continuing studies focusing on aberrant HDAC8 function in CdLS, we now report the preparation and biophysical evaluation of five human HDAC8 mutants: P91L, G117E, H180R, D233G, and G304R. Additionally, the double mutants D233G–Y306F and P91L–Y306F were prepared to enable cocrystallization of intact enzyme–substrate complexes. X-ray crystal structures of G117E, P91L–Y306F, and D233G–Y306F HDAC8 mutants reveal that each CdLS mutation causes structural changes that compromise catalysis and/or thermostability. For example, the D233G mutation disrupts the D233–K202–S276 hydrogen bond network, which stabilizes key tertiary structure interactions, thereby significantly compromising thermostability. Molecular dynamics simulations of H180R and G304R HDAC8 mutants suggest that the bulky arginine side chain of each mutant protrudes into the substrate binding site and also causes active site residue Y306 to fluctuate away from the position required for substrate activation and catalysis. Significantly, the catalytic activities of most mutants can be partially or fully rescued by the activator N-(phenylcarbamothioyl)-benzamide, suggesting that HDAC8 activators may serve as possible leads in the therapeutic management of CdLS.
Co-reporter:Golda G. Harris, Patrick M. Lombardi, Travis A. Pemberton, Tsutomu Matsui, Thomas M. Weiss, Kathryn E. Cole, Mustafa Köksal, Frank V. Murphy IV, L. Sangeetha Vedula, Wayne K. W. Chou, David E. Cane, and David W. Christianson
Biochemistry 2015 Volume 54(Issue 48) pp:7142-7155
Publication Date(Web):November 24, 2015
DOI:10.1021/acs.biochem.5b01143
Geosmin synthase from Streptomyces coelicolor (ScGS) catalyzes an unusual, metal-dependent terpenoid cyclization and fragmentation reaction sequence. Two distinct active sites are required for catalysis: the N-terminal domain catalyzes the ionization and cyclization of farnesyl diphosphate to form germacradienol and inorganic pyrophosphate (PPi), and the C-terminal domain catalyzes the protonation, cyclization, and fragmentation of germacradienol to form geosmin and acetone through a retro-Prins reaction. A unique αα domain architecture is predicted for ScGS based on amino acid sequence: each domain contains the metal-binding motifs typical of a class I terpenoid cyclase, and each domain requires Mg2+ for catalysis. Here, we report the X-ray crystal structure of the unliganded N-terminal domain of ScGS and the structure of its complex with three Mg2+ ions and alendronate. These structures highlight conformational changes required for active site closure and catalysis. Although neither full-length ScGS nor constructs of the C-terminal domain could be crystallized, homology models of the C-terminal domain were constructed on the basis of ∼36% sequence identity with the N-terminal domain. Small-angle X-ray scattering experiments yield low-resolution molecular envelopes into which the N-terminal domain crystal structure and the C-terminal domain homology model were fit, suggesting possible αα domain architectures as frameworks for bifunctional catalysis.
Co-reporter:Yang Hai, Eduard J. Kerkhoven, Michael P. Barrett, and David W. Christianson
Biochemistry 2015 Volume 54(Issue 2) pp:458-471
Publication Date(Web):December 23, 2014
DOI:10.1021/bi501366a
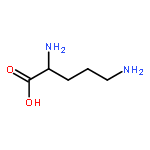
The X-ray crystal structure of an arginase-like protein from the parasitic protozoan Trypanosoma brucei, designated TbARG, is reported at 1.80 and 2.38 Å resolution in its reduced and oxidized forms, respectively. The oxidized form of TbARG is a disulfide-linked hexamer that retains the overall architecture of a dimer of trimers in the reduced form. Intriguingly, TbARG does not contain metal ions in its putative active site, and amino acid sequence comparisons indicate that all but one of the residues required for coordination to the catalytically obligatory binuclear manganese cluster in other arginases are substituted here with residues incapable of metal ion coordination. Therefore, the structure of TbARG is the first of a member of the arginase/deacetylase superfamily that is not a metalloprotein. Although we show that metal binding activity is easily reconstituted in TbARG by site-directed mutagenesis and confirmed in X-ray crystal structures, it is curious that this protein and its parasitic orthologues evolved away from metal binding function. Knockout of the TbARG gene from the genome demonstrated that its function is not essential to cultured bloodstream-form T. brucei, and metabolomics analysis confirmed that the enzyme has no role in the conversion of l-arginine to l-ornithine in these cells. While the molecular function of TbARG remains enigmatic, the fact that the T. brucei genome encodes only this protein and not a functional arginase indicates that the parasite must import l-ornithine from its host to provide a source of substrate for ornithine decarboxylase in the polyamine biosynthetic pathway, an active target for the development of antiparasitic drugs.
Co-reporter:Christophe Decroos, Christine M. Bowman, Joe-Ann S. Moser, Karen E. Christianson, Matthew A. Deardorff, and David W. Christianson
ACS Chemical Biology 2014 Volume 9(Issue 9) pp:2157
Publication Date(Web):July 30, 2014
DOI:10.1021/cb5003762
Cornelia de Lange Syndrome (CdLS) is a multiple congenital anomaly disorder resulting from mutations in genes that encode the core components of the cohesin complex, SMC1A, SMC3, and RAD21, or two of its regulatory proteins, NIPBL and HDAC8. HDAC8 is the human SMC3 lysine deacetylase required for cohesin recycling in the cell cycle. To date, 16 different missense mutations in HDAC8 have recently been identified in children diagnosed with CdLS. To understand the molecular effects of these mutations in causing CdLS and overlapping phenotypes, we have fully characterized the structure and function of five HDAC8 mutants: C153F, A188T, I243N, T311M, and H334R. X-ray crystal structures reveal that each mutation causes local structural changes that compromise catalysis and/or thermostability. For example, the C153F mutation triggers conformational changes that block acetate product release channels, resulting in only 2% residual catalytic activity. In contrast, the H334R mutation causes structural changes in a polypeptide loop distant from the active site and results in 91% residual activity, but the thermostability of this mutant is significantly compromised. Strikingly, the catalytic activity of these mutants can be partially or fully rescued in vitro by the HDAC8 activator N-(phenylcarbamothioyl)benzamide. These results suggest that HDAC8 activators might be useful leads in the search for new therapeutic strategies in managing CdLS.
Co-reporter:Ruiqiong Li, Wayne K. W. Chou, Julie A. Himmelberger, Kevin M. Litwin, Golda G. Harris, David E. Cane, and David W. Christianson
Biochemistry 2014 Volume 53(Issue 7) pp:
Publication Date(Web):February 11, 2014
DOI:10.1021/bi401643u
The class I terpenoid cyclase epi-isozizaene synthase (EIZS) utilizes the universal achiral isoprenoid substrate, farnesyl diphosphate, to generate epi-isozizaene as the predominant sesquiterpene cyclization product and at least five minor sesquiterpene products, making EIZS an ideal platform for the exploration of fidelity and promiscuity in a terpenoid cyclization reaction. The hydrophobic active site contour of EIZS serves as a template that enforces a single substrate conformation, and chaperones subsequently formed carbocation intermediates through a well-defined mechanistic sequence. Here, we have used the crystal structure of EIZS as a guide to systematically remold the hydrophobic active site contour in a library of 26 site-specific mutants. Remolded cyclization templates reprogram the reaction cascade not only by reproportioning products generated by the wild-type enzyme but also by generating completely new products of diverse structure. Specifically, we have tripled the overall number of characterized products generated by EIZS. Moreover, we have converted EIZS into six different sesquiterpene synthases: F96A EIZS is an (E)-β-farnesene synthase, F96W EIZS is a zizaene synthase, F95H EIZS is a β-curcumene synthase, F95M EIZS is a β-acoradiene synthase, F198L EIZS is a β-cedrene synthase, and F96V EIZS and W203F EIZS are (Z)-γ-bisabolene synthases. Active site aromatic residues appear to be hot spots for reprogramming the cyclization cascade by manipulating the stability and conformation of critical carbocation intermediates. A majority of mutant enzymes exhibit only relatively modest 2–100-fold losses of catalytic activity, suggesting that residues responsible for triggering substrate ionization readily tolerate mutations deeper in the active site cavity.
Co-reporter:Yang Hai, Jennifer E. Edwards, Michael C. Van Zandt, Karl F. Hoffmann, and David W. Christianson
Biochemistry 2014 Volume 53(Issue 28) pp:4671-4684
Publication Date(Web):July 9, 2014
DOI:10.1021/bi5004519
The X-ray crystal structure of arginase from Schistosoma mansoni (SmARG) and the structures of its complexes with several amino acid inhibitors have been determined at atomic resolution. SmARG is a binuclear manganese metalloenzyme that catalyzes the hydrolysis of l-arginine to form l-ornithine and urea, and this enzyme is upregulated in all forms of the parasite that interact with the human host. Current hypotheses suggest that parasitic arginases could play a role in host immune evasion by depleting pools of substrate l-arginine that would otherwise be utilized for NO biosynthesis and NO-dependent processes in the immune response. Although the amino acid sequence of SmARG is only 42% identical with that of human arginase I, residues important for substrate binding and catalysis are strictly conserved. In general, classical amino acid inhibitors such as 2(S)-amino-6-boronohexanoic acid (ABH) tend to bind more weakly to SmARG than to human arginase I despite identical inhibitor binding modes in each enzyme active site. The identification of a patch on the enzyme surface capable of accommodating the additional Cα substitutent of an α,α-disubstituted amino acid inhibitor suggests that such inhibitors could exhibit higher affinity and biological activity. The structures of SmARG complexed with two different α,α-disubstituted derivatives of ABH are presented and provide a proof of concept for this approach in the enhancement of enzyme–inhibitor affinity.
Co-reporter:Mengbin Chen, Naeemah Al-lami, Marine Janvier, Edward L. D’Antonio, Juan A. Faraldos, David E. Cane, Rudolf K. Allemann, and David W. Christianson
Biochemistry 2013 Volume 52(Issue 32) pp:
Publication Date(Web):August 1, 2013
DOI:10.1021/bi400691v
Aristolochene synthase, a metal-dependent sesquiterpene cyclase from Aspergillus terreus, catalyzes the ionization-dependent cyclization of farnesyl diphosphate (FPP) to form the bicyclic eremophilane (+)-aristolochene with perfect structural and stereochemical precision. Here, we report the X-ray crystal structure of aristolochene synthase complexed with three Mg2+ ions and the unreactive substrate analogue farnesyl-S-thiolodiphosphate (FSPP), showing that the substrate diphosphate group is anchored by metal coordination and hydrogen bond interactions identical to those previously observed in the complex with three Mg2+ ions and inorganic pyrophosphate (PPi). Moreover, the binding conformation of FSPP directly mimics that expected for productively bound FPP, with the exception of the precise alignment of the C–S bond with regard to the C10–C11 π system that would be required for C1–C10 bond formation in the first step of catalysis. We also report crystal structures of aristolochene synthase complexed with Mg2+3-PPi and ammonium or iminium analogues of bicyclic carbocation intermediates proposed for the natural cyclization cascade. Various binding orientations are observed for these bicyclic analogues, and these orientations appear to be driven by favorable electrostatic interactions between the positively charged ammonium group of the analogue and the negatively charged PPi anion. Surprisingly, the active site is sufficiently flexible to accommodate analogues with partially or completely incorrect stereochemistry. Although this permissiveness in binding is unanticipated, based on the stereochemical precision of catalysis that leads exclusively to the (+)-aristolochene stereoisomer, it suggests the ability of the active site to enable controlled reorientation of intermediates during the cyclization cascade. Taken together, these structures illuminate important aspects of the catalytic mechanism.
Co-reporter:Yang Hai, Reilly Jane Dugery, David Healy, and David W. Christianson
Biochemistry 2013 Volume 52(Issue 51) pp:
Publication Date(Web):November 21, 2013
DOI:10.1021/bi401352h
The crystal structure of formiminoglutamase from Trypanosoma cruzi (TcFIGase) is reported at 1.85 Å resolution. Although the structure of this enzyme was previously determined by the Structural Genomics of Pathogenic Protozoa Consortium (PDB accession code 2A0M), this structure was determined at low pH and lacked bound metal ions; accordingly, the protein was simply annotated as “arginase superfamily protein” with undetermined function. We show that reconstitution of this protein with Mn2+ confers maximal catalytic activity in the hydrolysis of formiminoglutamate to yield glutamate and formamide, thereby demonstrating that this protein is a metal-dependent formiminoglutamase. Equilibration of TcFIGase crystals with MnCl2 at higher pH yields a binuclear manganese cluster similar to that observed in arginase, except that the histidine ligand to the Mn2+A ion of arginase is an asparagine ligand (N114) to the Mn2+A ion of TcFIGase. The crystal structure of N114H TcFIGase reveals a binuclear manganese cluster essentially identical to that of arginase, but the mutant exhibits a modest 35% loss of catalytic efficiency (kcat/KM). Interestingly, when TcFIGase is prepared and crystallized in the absence of reducing agents at low pH, a disulfide linkage forms between C35 and C242 in the active site. When reconstituted with Mn2+ at higher pH, this oxidized enzyme exhibits a modest 33% loss of catalytic efficiency. Structure determinations of the metal-free and metal-bound forms of oxidized TcFIGase reveal that although disulfide formation constricts the main entrance to the active site, other structural changes open alternative channels to the active site that may help sustain catalytic activity.
Co-reporter:Christophe Decroos, Christine M. Bowman, David W. Christianson
Bioorganic & Medicinal Chemistry 2013 Volume 21(Issue 15) pp:4530-4540
Publication Date(Web):1 August 2013
DOI:10.1016/j.bmc.2013.05.045
Polyamines are small essential polycations involved in many biological processes. Enzymes of polyamine metabolism have been extensively studied and are attractive drug targets. Nevertheless, the reversible acetylation of polyamines remains poorly understood. Although eukaryotic N8-acetylspermidine deacetylase activity has already been detected and studied, the specific enzyme responsible for this activity has not yet been identified. However, a zinc deacetylase from Mycoplana ramosa, acetylpolyamine amidohydrolase (APAH), has been reported to use various acetylpolyamines as substrates. The recently solved crystal structure of this polyamine deacetylase revealed the formation of an ‘L’-shaped active site tunnel at the dimer interface, with ideal dimensions and electrostatic properties for accommodating narrow, flexible, cationic polyamine substrates. Here, we report the design, synthesis, and evaluation of N8-acetylspermidine analogues bearing different zinc binding groups as potential inhibitors of APAH. Most of the synthesized compounds exhibit modest potency, with IC50 values in the mid-micromolar range, but compounds bearing hydroxamate or trifluoromethylketone zinc binding groups exhibit enhanced inhibitory potency in the mid-nanomolar range. These inhibitors will enable future explorations of acetylpolyamine function in both prokaryotes and eukaryotes.
Co-reporter:Mustafa Köksal, Wayne K. W. Chou, David E. Cane, and David W. Christianson
Biochemistry 2013 Volume 52(Issue 31) pp:
Publication Date(Web):July 11, 2013
DOI:10.1021/bi400797c
The crystal structure of 2-methylisoborneol synthase (MIBS) from Streptomyces coelicolor A3(2) has been determined in its unliganded state and in complex with two Mg2+ ions and 2-fluoroneryl diphosphate at 1.85 and 2.00 Å resolution, respectively. Under normal circumstances, MIBS catalyzes the cyclization of the naturally occurring, noncanonical 11-carbon isoprenoid substrate, 2-methylgeranyl diphosphate, which first undergoes an ionization–isomerization–ionization sequence through the tertiary diphosphate intermediate 2-methyllinalyl diphosphate to enable subsequent cyclization chemistry. MIBS does not exhibit catalytic activity with 2-fluorogeranyl diphosphate, and we recently reported the crystal structure of MIBS complexed with this unreactive substrate analogue [Köksal, M., Chou, W. K. W., Cane, D. E., Christianson, D. W. (2012) Biochemistry51, 3011–3020]. However, cocrystallization of MIBS with the fluorinated analogue of the tertiary allylic diphosphate intermediate, 2-fluorolinalyl diphosphate, reveals unexpected reactivity for the intermediate analogue and yields the crystal structure of the complex with the primary allylic diphosphate, 2-fluoroneryl diphosphate. Comparison with the structure of the unliganded enzyme reveals that the crystalline enzyme active site remains partially open, presumably due to the binding of only two Mg2+ ions. Assays in solution indicate that MIBS catalyzes the generation of (1R)-(+)-camphor from the substrate 2-fluorolinalyl diphosphate, suggesting that both 2-fluorolinalyl diphosphate and 2-methyllinalyl diphosphate follow the identical cyclization mechanism leading to 2-substituted isoborneol products; however, the initially generated 2-fluoroisoborneol cyclization product is unstable and undergoes elimination of hydrogen fluoride to yield (1R)-(+)-camphor.
Co-reporter:Mustafa Köksal, Wayne K. W. Chou, David E. Cane, and David W. Christianson
Biochemistry 2012 Volume 51(Issue 14) pp:
Publication Date(Web):March 28, 2012
DOI:10.1021/bi201827a
The crystal structure of 2-methylisoborneol synthase (MIBS) from Streptomyces coelicolor A3(2) has been determined in complex with substrate analogues geranyl-S-thiolodiphosphate and 2-fluorogeranyl diphosphate at 1.80 and 1.95 Å resolution, respectively. This terpenoid cyclase catalyzes the cyclization of the naturally occurring, noncanonical C-methylated isoprenoid substrate, 2-methylgeranyl diphosphate, to form the bicyclic product 2-methylisoborneol, a volatile C11 homoterpene alcohol with an earthy, musty odor. While MIBS adopts the tertiary structure of a class I terpenoid cyclase, its dimeric quaternary structure differs from that previously observed in dimeric terpenoid cyclases from plants and fungi. The quaternary structure of MIBS is nonetheless similar in some respects to that of dimeric farnesyl diphosphate synthase, which is not a cyclase. The structures of MIBS complexed with substrate analogues provide insights regarding differences in the catalytic mechanism of MIBS and the mechanisms of (+)-bornyl diphosphate synthase and endo-fenchol synthase, plant cyclases that convert geranyl diphosphate into products with closely related bicyclic bornyl skeletons, but distinct structures and stereochemistries.
Co-reporter:Mustafa Köksal, Wayne K. W. Chou, David E. Cane, and David W. Christianson
Biochemistry 2012 Volume 51(Issue 14) pp:3003-3010
Publication Date(Web):March 28, 2012
DOI:10.1021/bi300109c
Geranyl diphosphate C-methyltransferase (GPPMT) from Streptomyces coelicolor A3(2) is the first methyltransferase discovered that modifies an acyclic isoprenoid diphosphate, geranyl diphosphate (GPP), to yield a noncanonical acyclic allylic diphosphate product, 2-methylgeranyl diphosphate, which serves as the substrate for a subsequent cyclization reaction catalyzed by a terpenoid cyclase, methylisoborneol synthase. Here, we report the crystal structures of GPPMT in complex with GPP or the substrate analogue geranyl S-thiolodiphosphate (GSPP) along with S-adenosyl-l-homocysteine in the cofactor binding site, resulting from in situ demethylation of S-adenosyl-l-methionine, at 2.05 or 1.82 Å resolution, respectively. These structures suggest that both GPP and GSPP can undergo catalytic methylation in crystalline GPPMT, followed by dissociation of the isoprenoid product. S-Adenosyl-l-homocysteine remains bound in the active site, however, and does not exchange with a fresh molecule of cofactor S-adenosyl-l-methionine. These structures provide important clues about the molecular mechanism of the reaction, especially with regard to the face of the 2,3 double bond of GPP that is methylated as well as the stabilization of the resulting carbocation intermediate through cation−π interactions.
Co-reporter:Edward L. D’Antonio, Yang Hai, and David W. Christianson
Biochemistry 2012 Volume 51(Issue 42) pp:
Publication Date(Web):October 12, 2012
DOI:10.1021/bi301145n
Various binuclear metal ion clusters and complexes have been reconstituted in crystalline human arginase I by removing the Mn2+2 cluster of the wild-type enzyme with metal chelators and subsequently soaking the crystalline apoenzyme in buffer solutions containing NiCl2 or ZnCl2. X-ray crystal structures of these metal ion variants are correlated with catalytic activity measurements that reveal differences resulting from metal ion substitution. Additionally, treatment of crystalline Mn2+2-human arginase I with Zn2+ reveals for the first time the structural basis for inhibition by Zn2+, which forms a carboxylate-histidine-Zn2+ triad with H141 and E277. The imidazole side chain of H141 is known to be hyper-reactive, and its chemical modification or mutagenesis is known to similarly compromise catalysis. The reactive substrate analogue 2(S)-amino-6-boronohexanoic acid (ABH) binds as a tetrahedral boronate anion to Mn2+2, Co2+2, Ni2+2, and Zn2+2 clusters in human arginase I, and it can be stabilized by a third inhibitory Zn2+ ion coordinated by H141. Because ABH binds as an analogue of the tetrahedral intermediate and its flanking transition states in catalysis, this implies that the various metallo-substituted enzymes are capable of some level of catalysis with an actual substrate. Accordingly, we establish the following trend for turnover number (kcat) and catalytic efficiency (kcat/KM): Mn2+ > Ni2+ ≈ Co2+ ≫ Zn2+. Therefore, Mn2+ is required for optimal catalysis by human arginase I.
Co-reporter:Mo Chen, Adegoke O. Adeniji, Barry M. Twenter, Jeffrey D. Winkler, David W. Christianson, Trevor M. Penning
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 10) pp:3492-3497
Publication Date(Web):15 May 2012
DOI:10.1016/j.bmcl.2012.03.085
Castrate resistant prostate cancer (CRPC) is associated with increased androgen receptor (AR) signaling often brought about by elevated intratumoral androgen biosynthesis and AR amplification. Inhibition of androgen biosynthesis and/or AR antagonism should be efficacious in the treatment of CRPC. AKR1C3 catalyzes the formation of potent AR ligands from inactive precursors and is one of the most upregulated genes in CRPC. AKR1C3 inhibitors should not inhibit the related isoforms, AKR1C1 and AKR1C2 that are involved in 5α-dihydrotestosterone inactivation in the prostate. We have previously developed a series of flufenamic acid analogs as potent and selective AKR1C3 inhibitors [Adeniji, A. O. et al., J. Med. Chem.2012, 55, 2311]. Here we report the X-ray crystal structure of one lead compound 3-((4-(trifluoromethyl)phenyl) amino)benzoic acid (1) in complex with AKR1C3. Compound 1 adopts a similar binding orientation as flufenamic acid, however, its phenylamino ring projects deeper into a subpocket and confers selectivity over the other AKR1C isoforms. We exploited the observation that some flufenamic acid analogs also act as AR antagonists and synthesized a second generation inhibitor, 3-((4-nitronaphthalen-1-yl)amino)benzoic acid (2). Compound 2 retained nanomolar potency and selective inhibition of AKR1C3 but also acted as an AR antagonist. It inhibited 5α-dihydrotestosterone stimulated AR reporter gene activity with an IC50 = 4.7 μM and produced a concentration dependent reduction in androgen receptor levels in prostate cancer cells. The in vitro and cell-based effects of compound 2 make it a promising lead for development of dual acting agent for CRPC. To illuminate the structural basis of AKR1C3 inhibition, we also report the crystal structure of the AKR1C3·NADP+·2 complex, which shows that compound 2 forms a unique double-decker structure with AKR1C3.Superimposed structures of AKR1C3 in complex with two N-aryl(amino)benzoic acid analogs, compound 1 (magenta) and compound 2 (blue).
Co-reporter:Kathryn E. Cole ; Daniel P. Dowling ; Matthew A. Boone ; Andrew J. Phillips
Journal of the American Chemical Society 2011 Volume 133(Issue 32) pp:12474-12477
Publication Date(Web):July 26, 2011
DOI:10.1021/ja205972n
Largazole is a macrocyclic depsipeptide originally isolated from the marine cyanobacterium Symploca sp., which is indigenous to the warm, blue-green waters of Key Largo, Florida (whence largazole derives its name). Largazole contains an unusual thiazoline–thiazole ring system that rigidifies its macrocyclic skeleton, and it also contains a lipophilic thioester side chain. Hydrolysis of the thioester in vivo yields largazole thiol, which exhibits remarkable antiproliferative effects and is believed to be the most potent inhibitor of the metal-dependent histone deacetylases (HDACs). Here, the 2.14 Å-resolution crystal structure of the HDAC8–largazole thiol complex is the first of an HDAC complexed with a macrocyclic inhibitor and reveals that ideal thiolate–zinc coordination geometry is the key chemical feature responsible for its exceptional affinity and biological activity. Notably, the core structure of largazole is conserved in romidepsin, a depsipeptide natural product formulated as the drug Istodax recently approved for cancer chemotherapy. Accordingly, the structure of the HDAC8–largazole thiol complex is the first to illustrate the mode of action of a new class of therapeutically important HDAC inhibitors.
Co-reporter:Monica Ilies ; Luigi Di Costanzo ; Daniel P. Dowling ; Katherine J. Thorn
Journal of Medicinal Chemistry 2011 Volume 54(Issue 15) pp:5432-5443
Publication Date(Web):July 5, 2011
DOI:10.1021/jm200443b
Arginase is a binuclear manganese metalloenzyme that hydrolyzes l-arginine to form l-ornithine and urea, and aberrant arginase activity is implicated in various diseases such as erectile dysfunction, asthma, atherosclerosis, and cerebral malaria. Accordingly, arginase inhibitors may be therapeutically useful. Continuing our efforts to expand the chemical space of arginase inhibitor design and inspired by the binding of 2-(difluoromethyl)-l-ornithine to human arginase I, we now report the first study of the binding of α,α-disubstituted amino acids to arginase. Specifically, we report the design, synthesis, and assay of racemic 2-amino-6-borono-2-methylhexanoic acid and racemic 2-amino-6-borono-2-(difluoromethyl)hexanoic acid. X-ray crystal structures of human arginase I and Plasmodium falciparum arginase complexed with these inhibitors reveal the exclusive binding of the l-stereoisomer; the additional α-substituent of each inhibitor is readily accommodated and makes new intermolecular interactions in the outer active site of each enzyme. Therefore, this work highlights a new region of the protein surface that can be targeted for additional affinity interactions, as well as the first comparative structural insights on inhibitor discrimination between a human and a parasitic arginase.
Co-reporter:Edward L. D’Antonio and David W. Christianson
Biochemistry 2011 Volume 50(Issue 37) pp:
Publication Date(Web):August 26, 2011
DOI:10.1021/bi201101t
The binuclear manganese metalloenzyme human arginase I (HAI) is a potential protein drug for cancer chemotherapy, in that it is capable of depleting extracellular l-Arg levels in the microenvironment of tumor cells that require this nutrient to thrive. Substitution of the native Mn2+2 cluster with a Co2+2 cluster in the active site yields an enzyme with enhanced catalytic activity at physiological pH (∼7.4) that could serve as an improved protein drug for l-Arg depletion therapy [Stone, E. M., Glazer, E. S., Chantranupong, L., Cherukuri, P., Breece, R. M., Tierney, D. L., Curley, S. A., Iverson, B. L., and Georgiou, G. (2010) ACS Chem. Biol. 5, 333–342]. A different catalytic mechanism is proposed for Co2+2-HAI compared with that of Mn2+2-HAI, including an unusual Nε–Co2+ coordination mode, to rationalize the lower KM value of l-Arg and the lower Ki value of l-Orn. However, we now report that no unusual metal coordination modes are observed in the cobalt-reconstituted enzyme. The X-ray crystal structures of unliganded Co2+2-HAI determined at 2.10 Å resolution (pH 7.0) and 1.97 Å resolution (pH 8.5), as well as the structures of Co2+2-HAI complexed with the reactive substrate analogue 2(S)-amino-6-boronohexanoic acid (ABH, pH 7.0) and the catalytic product l-Orn (pH 7.0) determined at 1.85 and 1.50 Å resolution, respectively, are essentially identical to the corresponding structures of Mn2+2-HAI. Therefore, in the absence of significant structural differences between Co2+2-HAI and Mn2+2-HAI, we suggest that a higher concentration of metal-bridging hydroxide ion at physiological pH for Co2+2-HAI, a consequence of the lower pKa of a Co2+-bound water molecule compared with a Mn2+-bound water molecule, strengthens electrostatic interactions with cationic amino acids and accounts for enhanced affinity as reflected in the lower KM value of l-Arg and the lower Ki value of l-Orn.
Co-reporter:Kathryn E. Cole, Samuel G. Gattis, Heather D. Angell, Carol A. Fierke, and David W. Christianson
Biochemistry 2011 Volume 50(Issue 2) pp:
Publication Date(Web):December 20, 2010
DOI:10.1021/bi101622a
The first committed step of lipid A biosynthesis is catalyzed by UDP-(3-O-((R)-3-hydroxymyristoyl))-N-acetylglucosamine deacetylase, a metal-dependent deacetylase also known as LpxC. Because lipid A is essential for bacterial viability, the inhibition of LpxC is an appealing therapeutic strategy for the treatment of Gram-negative bacterial infections. Here we report the 1.79 Å resolution X-ray crystal structure of LpxC from Yersinia enterocolitica (YeLpxC) complexed with the potent hydroxamate inhibitor CHIR-090. This enzyme is a nearly identical orthologue of LpxC from Yersinia pestis (99.7% sequence identity), the pathogen that causes bubonic plague. Similar to the inhibition of LpxC from Escherichia coli, CHIR-090 inhibits YeLpxC via a two-step slow, tight-binding mechanism with an apparent Ki of 0.54 ± 0.14 nM followed by conversion of the E·I to E·I* species with a rate constant of 0.11 ± 0.01 min−1. The structure of the LpxC complex with CHIR-090 shows that the inhibitor hydroxamate group chelates the active site zinc ion, and the “tail” of the inhibitor binds in the hydrophobic tunnel in the active site. This hydrophobic tunnel is framed by a βαβ subdomain that exhibits significant conformational flexibility as it accommodates inhibitor binding. CHIR-090 displays a 27 mm zone of inhibition against Y. enterocolitica in a Kirby−Bauer antibiotic assay, which is comparable to its reported activity against other Gram-negative species including E. coli and Pseudomonas aeruginosa. This study demonstrates that the inhibition of LpxC should be explored as a potential therapeutic and/or prophylatic response to infection by weaponized Yersinia species.
Co-reporter:Patrick M. Lombardi, Heather D. Angell, Douglas A. Whittington, Erin F. Flynn, Kanagalaghatta R. Rajashankar, and David W. Christianson
Biochemistry 2011 Volume 50(Issue 11) pp:1808-1817
Publication Date(Web):January 26, 2011
DOI:10.1021/bi101859k
Polyamines are a ubiquitous class of polycationic small molecules that can influence gene expression by binding to nucleic acids. Reversible polyamine acetylation regulates nucleic acid binding and is required for normal cell cycle progression and proliferation. Here, we report the structures of Mycoplana ramosa acetylpolyamine amidohydrolase (APAH) complexed with a transition state analogue and a hydroxamate inhibitor and an inactive mutant complexed with two acetylpolyamine substrates. The structure of APAH is the first of a histone deacetylase-like oligomer and reveals that an 18-residue insert in the L2 loop promotes dimerization and the formation of an 18 Å long “L”-shaped active site tunnel at the dimer interface, accessible only to narrow and flexible substrates. The importance of dimerization for polyamine deacetylase function leads to the suggestion that a comparable dimeric or double-domain histone deacetylase could catalyze polyamine deacetylation reactions in eukaryotes.
Co-reporter:Monica Ilies, Daniel P. Dowling, Patrick M. Lombardi, David W. Christianson
Bioorganic & Medicinal Chemistry Letters 2011 21(19) pp: 5854-5858
Publication Date(Web):
DOI:10.1016/j.bmcl.2011.07.100
Co-reporter:Monica Ilies ; Luigi Di Costanzo ; Michelle L. North ; Jeremy A. Scott
Journal of Medicinal Chemistry 2010 Volume 53(Issue 10) pp:4266-4276
Publication Date(Web):May 4, 2010
DOI:10.1021/jm100306a
Arginase, a key metalloenzyme of the urea cycle that converts l-arginine into l-ornithine and urea, is presently considered a pharmaceutical target for the management of diseases associated with aberrant l-arginine homeostasis, such as asthma, cardiovascular diseases, and erectile dysfunction. We now report the design, synthesis, and evaluation of a series of 2-aminoimidazole amino acid inhibitors in which the 2-aminoimidazole moiety serves as a guanidine mimetic. These compounds represent a new class of arginase inhibitors. The most potent inhibitor identified in this study, 2-(S)-amino-5-(2-aminoimidazol-1-yl)pentanoic acid (A1P, 10), binds to human arginase I with Kd = 2 μM and significantly attenuates airways hyperresponsiveness in a murine model of allergic airways inflammation. These findings suggest that 2-aminoimidazole amino acids represent new leads for the development of arginase inhibitors with promising pharmacological profiles.
Co-reporter:Julie A. Aaron, Xin Lin, David E. Cane and David W. Christianson
Biochemistry 2010 Volume 49(Issue 8) pp:
Publication Date(Web):February 4, 2010
DOI:10.1021/bi902088z
The X-ray crystal structure of recombinant epi-isozizaene synthase (EIZS), a sesquiterpene cyclase from Streptomyces coelicolor A3(2), has been determined at 1.60 Å resolution. Specifically, the structure of wild-type EIZS is that of its closed conformation in complex with three Mg2+ ions, inorganic pyrophosphate (PPi), and the benzyltriethylammonium cation (BTAC). Additionally, the structure of D99N EIZS has been determined in an open, ligand-free conformation at 1.90 Å resolution. Comparison of these two structures provides the first view of conformational changes required for substrate binding and catalysis in a bacterial terpenoid cyclase. Moreover, the binding interactions of BTAC may mimic those of a carbocation intermediate in catalysis. Accordingly, the aromatic rings of F95, F96, and F198 appear to be well-oriented to stabilize carbocation intermediates in the cyclization cascade through cation−π interactions. Mutagenesis of aromatic residues in the enzyme active site results in the production of alternative sesquiterpene product arrays due to altered modes of stabilization of carbocation intermediates as well as altered templates for the cyclization of farnesyl diphosphate. Accordingly, the 1.64 Å resolution crystal structure of F198A EIZS in a complex with three Mg2+ ions, PPi, and BTAC reveals an alternative binding orientation of BTAC; alternative binding orientations of a carbocation intermediate could lead to the formation of alternative products. Finally, the crystal structure of wild-type EIZS in a complex with four Hg2+ ions has been determined at 1.90 Å resolution, showing that metal binding triggers a significant conformational change of helix G to cap the active site.
Co-reporter:Daniel P. Dowling, Samuel G. Gattis, Carol A. Fierke and David W. Christianson
Biochemistry 2010 Volume 49(Issue 24) pp:
Publication Date(Web):May 27, 2010
DOI:10.1021/bi1005046
The metal-dependent histone deacetylases (HDACs) adopt an α/β protein fold first identified in rat liver arginase. Despite insignificant overall amino acid sequence identity, these enzymes share a strictly conserved metal binding site with divergent metal specificity and stoichiometry. HDAC8, originally thought to be a Zn2+-metallohydrolase, exhibits increased activity with Co2+ and Fe2+ cofactors based on kcat/KM (Gantt, S. L., Gattis, S. G., and Fierke, C. A. (2006) Biochemistry 45, 6170−6178). Here, we report the first X-ray crystal structures of metallo-substituted HDAC8, Co2+-HDAC8, D101L Co2+-HDAC8, D101L Mn2+-HDAC8, and D101L Fe2+-HDAC8, each complexed with the inhibitor M344. Metal content of protein samples in solution is confirmed by inductively coupled plasma mass spectrometry. For the crystalline enzymes, peaks in Bijvoet difference Fourier maps calculated from X-ray diffraction data collected near the respective elemental absorption edges confirm metal substitution. Additional solution studies confirm incorporation of Cu2+; Fe3+ and Ni2+ do not bind under conditions tested. The metal dependence of the substrate KM values and the Ki values of hydroxamate inhibitors that chelate the active site metal are consistent with substrate−metal coordination in the precatalytic Michaelis complex that enhances catalysis. Additionally, although HDAC8 binds Zn2+ nearly 106-fold more tightly than Fe2+, the affinities for both metal ions are comparable to the readily exchangeable metal concentrations estimated in living cells, suggesting that HDAC8 could bind either or both Fe2+ or Zn2+ in vivo.
Co-reporter:Daniel P. Dowling, Monica Ilies, Kellen L. Olszewski, Silvia Portugal, Maria M. Mota, Manuel Llinás and David W. Christianson
Biochemistry 2010 Volume 49(Issue 26) pp:
Publication Date(Web):June 9, 2010
DOI:10.1021/bi100390z
The 2.15 Å resolution crystal structure of arginase from Plasmodium falciparum, the parasite that causes cerebral malaria, is reported in complex with the boronic acid inhibitor 2(S)-amino-6-boronohexanoic acid (ABH) (Kd = 11 μM). This is the first crystal structure of a parasitic arginase. Various protein constructs were explored to identify an optimally active enzyme form for inhibition and structural studies and to probe the structure and function of two polypeptide insertions unique to malarial arginase: a 74-residue low-complexity region contained in loop L2 and an 11-residue segment contained in loop L8. Structural studies indicate that the low-complexity region is largely disordered and is oriented away from the trimer interface; its deletion does not significantly compromise enzyme activity. The loop L8 insertion is located at the trimer interface and makes several intra- and intermolecular interactions important for enzyme function. In addition, we also demonstrate that arg- Plasmodium berghei sporozoites show significantly decreased liver infectivity in vivo. Therefore, inhibition of malarial arginase may serve as a possible candidate for antimalarial therapy against liver-stage infection, and ABH may serve as a lead for the development of inhibitors.
Co-reporter:Tatiana Y. Zakharian, David W. Christianson
Tetrahedron Letters 2010 Volume 51(Issue 28) pp:3645-3648
Publication Date(Web):14 July 2010
DOI:10.1016/j.tetlet.2010.05.017
Synthesis of C60–benzenesulfonamide conjugates is reported. The strategies for improving their water solubility, as required for binding to human carbonic anhydrase II, are discussed.Synthesis of C60–benzenesulfonamide conjugates is reported. The strategies for improving their water solubility, as required for binding to human carbonic anhydrase II, are discussed.
Co-reporter:Heather A. Gennadios, Veronica Gonzalez, Luigi Di Costanzo, Amang Li, Fanglei Yu, David J. Miller, Rudolf K. Allemann and David W. Christianson
Biochemistry 2009 Volume 48(Issue 26) pp:
Publication Date(Web):June 2, 2009
DOI:10.1021/bi900483b
(+)-δ-Cadinene synthase (DCS) from Gossypium arboreum (tree cotton) is a sesquiterpene cyclase that catalyzes the cyclization of farnesyl diphosphate in the first committed step of the biosynthesis of gossypol, a phytoalexin that defends the plant from bacterial and fungal pathogens. Here, we report the X-ray crystal structure of unliganded DCS at 2.4 Å resolution and the structure of its complex with three putative Mg2+ ions and the substrate analogue inhibitor 2-fluorofarnesyl diphosphate (2F-FPP) at 2.75 Å resolution. These structures illuminate unusual features that accommodate the trinuclear metal cluster required for substrate binding and catalysis. Like other terpenoid cyclases, DCS contains a characteristic aspartate-rich D307DTYD311 motif on helix D that interacts with Mg2+A and Mg2+C. However, DCS appears to be unique among terpenoid cyclases in that it does not contain the “NSE/DTE” motif on helix H that specifically chelates Mg2+B, which is usually found as the signature sequence (N,D)D(L,I,V)X(S,T)XXXE (boldface indicates Mg2+B ligands). Instead, DCS contains a second aspartate-rich motif, D451DVAE455, that interacts with Mg2+B. In this regard, DCS is more similar to the isoprenoid chain elongation enzyme farnesyl diphosphate synthase, which also contains two aspartate-rich motifs, rather than the greater family of terpenoid cyclases. Nevertheless, the structure of the DCS−2F-FPP complex shows that the structure of the trinuclear magnesium cluster is generally similar to that of other terpenoid cyclases despite the alternative Mg2+B binding motif. Analyses of DCS mutants with alanine substitutions in the D307DTYD311 and D451DVAE455 segments reveal the contributions of these segments to catalysis.
Co-reporter:Ekaterina Y. Shishova, Luigi Di Costanzo, Francis A. Emig, David E. Ash and David W. Christianson
Biochemistry 2009 Volume 48(Issue 1) pp:
Publication Date(Web):December 18, 2008
DOI:10.1021/bi801911v
Arginase is a binuclear manganese metalloenzyme that serves as a therapeutic target for the treatment of asthma, erectile dysfunction, and atherosclerosis. In order to better understand the molecular basis of inhibitor affinity, we have employed site-directed mutagenesis, enzyme kinetics, and X-ray crystallography to probe the molecular recognition of the amino acid moiety (i.e., the α-amino and α-carboxylate groups) of substrate l-arginine and inhibitors in the active site of arginase I. Specifically, we focus on (1) a water-mediated hydrogen bond between the substrate α-carboxylate and T135, (2) a direct hydrogen bond between the substrate α-carboxylate and N130, and (3) a direct charged hydrogen bond between the substrate α-amino group and D183. Amino acid substitutions for T135, N130, and D183 generally compromise substrate affinity as reflected by increased KM values but have less pronounced effects on catalytic function as reflected by minimal variations of kcat. As with substrate KM values, inhibitor Kd values increase for binding to enzyme mutants and suggest that the relative contribution of intermolecular interactions to amino acid affinity in the arginase active site is water-mediated hydrogen bond < direct hydrogen bond < direct charged hydrogen bond. Structural comparisons of arginase with the related binuclear manganese metalloenzymes agmatinase and proclavaminic acid amidinohydrolase suggest that the evolution of substrate recognition in the arginase fold occurs by mutation of residues contained in specificity loops flanking the mouth of the active site (especially loops 4 and 5), thereby allowing diverse guanidinium substrates to be accommodated for catalysis.
Co-reporter:Tatiana Y. Zakharian, Luigi Di Costanzo and David W. Christianson
Organic & Biomolecular Chemistry 2008 vol. 6(Issue 18) pp:3240-3243
Publication Date(Web):06 Aug 2008
DOI:10.1039/B811797G
The synthesis of (2S)-2-amino-7,8-epoxyoctanoic acid is reported along with the X-ray crystal structure of its complex with human arginase I, revealing unique coordination interactions with two manganese ions in the enzyme active site.
Co-reporter:Daniel P. Dowling, Stephanie L. Gantt, Samuel G. Gattis, Carol A. Fierke and David W. Christianson
Biochemistry 2008 Volume 47(Issue 51) pp:13554-13563
Publication Date(Web):November 24, 2008
DOI:10.1021/bi801610c
Metal-dependent histone deacetylases (HDACs) require Zn2+ or Fe2+ to regulate the acetylation of lysine residues in histones and other proteins in eukaryotic cells. Isozyme HDAC8 is perhaps the archetypical member of the class I HDAC family and serves as a paradigm for studying structure−function relationships. Here, we report the structures of HDAC8 complexes with trichostatin A and 3-(1-methyl-4-phenylacetyl-1H-2-pyrrolyl)-N-hydroxy-2-propenamide (APHA) in a new crystal form. The structure of the APHA complex reveals that the hydroxamate C═O group accepts a hydrogen bond from Y306 but does not coordinate to Zn2+ with favorable geometry, perhaps due to the constraints of its extended π system. Additionally, since APHA binds to only two of the three protein molecules in the asymmetric unit of this complex, the structure of the third monomer represents the first structure of HDAC8 in the unliganded state. Comparison of unliganded and liganded structures illustrates ligand-induced conformational changes in the L2 loop that likely accompany substrate binding and catalysis. Furthermore, these structures, along with those of the D101N, D101E, D101A, and D101L variants, support the proposal that D101 is critical for the function of the L2 loop. However, amino acid substitutions for D101 can also trigger conformational changes of Y111 and W141 that perturb the substrate binding site. Finally, the structure of H143A HDAC8 complexed with an intact acetylated tetrapeptide substrate molecule confirms the importance of D101 for substrate binding and reveals how Y306 and the active site zinc ion together bind and activate the scissile amide linkage of acetyllysine.
Co-reporter:D. P. Dowling;L. Di Costanzo;H. A. Gennadios
Cellular and Molecular Life Sciences 2008 Volume 65( Issue 13) pp:2039-2055
Publication Date(Web):2008 July
DOI:10.1007/s00018-008-7554-z
Novel structural superfamilies can be identified among the large number of protein structures deposited in the Protein Data Bank based on conservation of fold in addition to conservation of amino acid sequence. Since sequence diverges more rapidly than fold in protein Evolution, proteins with little or no significant sequence identity are occasionally observed to adopt similar folds, thereby reflecting unanticipated evolutionary relationships. Here, we review the unique α/β fold first observed in the manganese metalloenzyme rat liver arginase, consisting of a parallel eight-stranded β-sheet surrounded by several helices, and its evolutionary relationship with the zinc-requiring and/or iron-requiring histone deacetylases and acetylpolyamine amidohydrolases. Structural comparisons reveal key features of the core α/β fold that contribute to the divergent metal ion specificity and stoichiometry required for the chemical and biological functions of these enzymes.
Co-reporter:Hyunshun Shin, Heather A. Gennadios, Douglas A. Whittington, David W. Christianson
Bioorganic & Medicinal Chemistry 2007 Volume 15(Issue 7) pp:2617-2623
Publication Date(Web):1 April 2007
DOI:10.1016/j.bmc.2007.01.044
The first committed step in lipid A biosynthesis is catalyzed by uridine diphosphate-(3-O-(R-3-hydroxymyristoyl))-N-acetylglucosamine deacetylase (LpxC), a zinc-dependent deacetylase, and inhibitors of LpxC may be useful in the development of antibacterial agents targeting a broad spectrum of Gram-negative bacteria. Here, we report the design of amphipathic benzoic acid derivatives that bind in the hydrophobic tunnel in the active site of LpxC. The hydrophobic tunnel accounts for the specificity of LpxC toward substrates and substrate analogues bearing a 3-O-myristoyl substituent. Simple benzoic acid derivatives bearing an aliphatic ‘tail’ bind in the hydrophobic tunnel with micromolar affinity despite the lack of a glucosamine ring like that of the substrate. However, although these benzoic acid derivatives each contain a negatively charged carboxylate ‘warhead’ intended to coordinate to the active site zinc ion, the 2.25 Å resolution X-ray crystal structure of LpxC complexed with 3-(heptyloxy)benzoate reveals ‘backward’ binding in the hydrophobic tunnel, such that the benzoate moiety does not coordinate to zinc. Instead, it binds at the outer end of the hydrophobic tunnel. Interestingly, these ligands bind with affinities comparable to those measured for more complicated substrate analogue inhibitors containing glucosamine ring analogues and hydroxamate ‘warheads’ that coordinate to the active site zinc ion. We conclude that the intermolecular interactions in the hydrophobic tunnel dominate enzyme affinity in this series of benzoic acid derivatives.Benzoic acid derivatives bearing aliphatic substituents bind to LpxC with micromolar affinity. Surprisingly, the X-ray crystal structure of the complex with 3-(heptyloxy)benzoate reveals a ‘backward’ binding mode.
Co-reporter:David W Christianson, Nigel S Scrutton
Current Opinion in Structural Biology (December 2016) Volume 41() pp:viii-x
Publication Date(Web):1 December 2016
DOI:10.1016/j.sbi.2016.09.005
Co-reporter:Mustafa Köksal, Ina Zimmer, Jörg-Peter Schnitzler, David W. Christianson
Journal of Molecular Biology (17 September 2010) Volume 402(Issue 2) pp:363-373
Publication Date(Web):17 September 2010
DOI:10.1016/j.jmb.2010.07.009
The X-ray crystal structure of recombinant PcISPS (isoprene synthase from gray poplar hybrid Populus × canescens) has been determined at 2.7 Å resolution, and the structure of its complex with three Mg2+ and the unreactive substrate analogue dimethylallyl-S-thiolodiphosphate has been determined at 2.8 Å resolution. Analysis of these structures suggests that the generation of isoprene from substrate dimethylallyl diphosphate occurs via a syn-periplanar elimination mechanism in which the diphosphate-leaving group serves as a general base. This chemical mechanism is responsible for the annual atmospheric emission of 100 Tg of isoprene by terrestrial plant life. Importantly, the PcISPS structure promises to guide future protein engineering studies, potentially leading to hydrocarbon fuels and products that do not rely on traditional petrochemical sources.
Co-reporter:Luigi Di Costanzo, German A. Gomez, David W. Christianson
Journal of Molecular Biology (16 February 2007) Volume 366(Issue 2) pp:481-493
Publication Date(Web):16 February 2007
DOI:10.1016/j.jmb.2006.11.023
Aldehyde dehydrogenases catalyze the oxidation of aldehyde substrates to the corresponding carboxylic acids. Lactaldehyde dehydrogenase from Escherichia coli (aldA gene product, P25553) is an NAD+-dependent enzyme implicated in the metabolism of l-fucose and l-rhamnose. During the heterologous expression and purification of taxadiene synthase from the Pacific yew, lactaldehyde dehydrogenase from E. coli was identified as a minor (≤ 5%) side-product subsequent to its unexpected crystallization. Accordingly, we now report the serendipitous crystal structure determination of unliganded lactaldehyde dehydrogenase from E. coli determined by the technique of multiple isomorphous replacement using anomalous scattering at 2.2 Å resolution. Additionally, we report the crystal structure of the ternary enzyme complex with products lactate and NADH at 2.1 Å resolution, and the crystal structure of the enzyme complex with NADPH at 2.7 Å resolution. The structure of the ternary complex reveals that the nicotinamide ring of the cofactor is disordered between two conformations: one with the ring positioned in the active site in the so-called hydrolysis conformation, and another with the ring extended out of the active site into the solvent region, designated the out conformation. This represents the first crystal structure of an aldehyde dehydrogenase-product complex. The active site pocket in which lactate binds is more constricted than that of medium-chain dehydrogenases such as the YdcW gene product of E. coli. The structure of the binary complex with NADPH reveals the first view of the structural basis of specificity for NADH: the negatively charged carboxylate group of E179 destabilizes the binding of the 2′-phosphate group of NADPH sterically and electrostatically, thereby accounting for the lack of enzyme activity with this cofactor.
Co-reporter:L. Sangeetha Vedula, Yuxin Zhao, Robert M. Coates, Tanetoshi Koyama, David E. Cane, David W. Christianson
Archives of Biochemistry and Biophysics (15 October 2007) Volume 466(Issue 2) pp:
Publication Date(Web):15 October 2007
DOI:10.1016/j.abb.2007.06.016
Trichodiene synthase is a terpenoid cyclase that catalyzes the cyclization of farnesyl diphosphate (FPP) to form the bicyclic sesquiterpene hydrocarbon trichodiene (89%), at least five sesquiterpene side products (11%), and inorganic pyrophosphate (PPi). Incubation of trichodiene synthase with 2-fluorofarnesyl diphosphate or 4-methylfarnesyl diphosphate similarly yields sesquiterpene mixtures despite the electronic effects or steric bulk introduced by substrate derivatization. The versatility of the enzyme is also demonstrated in the 2.85 Å resolution X-ray crystal structure of the complex with Mg2+3-PPi and the benzyl triethylammonium cation, which is a bulkier mimic of the bisabolyl carbocation intermediate in catalysis. Taken together, these findings show that the active site of trichodiene synthase is sufficiently flexible to accommodate bulkier and electronically-diverse substrates and intermediates, which could indicate additional potential for the biosynthetic utility of this terpenoid cyclase.
Co-reporter:Edward L. D’Antonio, Buddy Ullman, Sigrid C. Roberts, Upasna Gaur Dixit, Mary E. Wilson, Yang Hai, David W. Christianson
Archives of Biochemistry and Biophysics (15 July 2013) Volume 535(Issue 2) pp:163-176
Publication Date(Web):15 July 2013
DOI:10.1016/j.abb.2013.03.015
Co-reporter:L. Sangeetha Vedula, Jiaoyang Jiang, Tatiana Zakharian, David E. Cane, David W. Christianson
Archives of Biochemistry and Biophysics (15 January 2008) Volume 469(Issue 2) pp:
Publication Date(Web):15 January 2008
DOI:10.1016/j.abb.2007.10.015
Trichodiene synthase from Fusarium sporotrichioides contains two metal ion-binding motifs required for the cyclization of farnesyl diphosphate: the “aspartate-rich” motif D100DXX(D/E) that coordinates to Mg2+A and Mg2+C, and the “NSE/DTE” motif N225DXXSXXXE that chelates Mg2+B (boldface indicates metal ion ligands). Here, we report steady-state kinetic parameters, product array analyses, and X-ray crystal structures of trichodiene synthase mutants in which the fungal NSE motif is progressively converted into a plant-like DDXXTXXXE motif, resulting in a degradation in both steady-state kinetic parameters and product specificity. Each catalytically active mutant generates a different distribution of sesquiterpene products, and three newly detected sesquiterpenes are identified. In addition, the kinetic and structural properties of the Y295F mutant of trichodiene synthase were found to be similar to those of the wild-type enzyme, thereby ruling out a proposed role for Y295 in catalysis.
Co-reporter:Luigi Di Costanzo, Martine Moulin, Michael Haertlein, Flora Meilleur, David W. Christianson
Archives of Biochemistry and Biophysics (1 September 2007) Volume 465(Issue 1) pp:
Publication Date(Web):1 September 2007
DOI:10.1016/j.abb.2007.04.036
Arginase is a manganese metalloenzyme that catalyzes the hydrolysis of l-arginine to yield l-ornithine and urea. In order to establish a foundation for future neutron diffraction studies that will provide conclusive structural information regarding proton/deuteron positions in enzyme–inhibitor complexes, we have expressed, purified, assayed, and determined the X-ray crystal structure of perdeuterated (i.e., fully deuterated) human arginase I complexed with 2(S)-amino-6-boronohexanoic acid (ABH) at 1.90 Å resolution. Prior to the neutron diffraction experiment, it is important to establish that perdeuteration does not cause any unanticipated structural or functional changes. Accordingly, we find that perdeuterated human arginase I exhibits catalytic activity essentially identical to that of the unlabeled enzyme. Additionally, the structure of the perdeuterated human arginase I–ABH complex is identical to that of the corresponding complex with the unlabeled enzyme. Therefore, we conclude that crystals of the perdeuterated human arginase I-ABH complex are suitable for neutron crystallographic study.
Co-reporter:Tatiana Y. Zakharian, Luigi Di Costanzo and David W. Christianson
Organic & Biomolecular Chemistry 2008 - vol. 6(Issue 18) pp:NaN3243-3243
Publication Date(Web):2008/08/06
DOI:10.1039/B811797G
The synthesis of (2S)-2-amino-7,8-epoxyoctanoic acid is reported along with the X-ray crystal structure of its complex with human arginase I, revealing unique coordination interactions with two manganese ions in the enzyme active site.









![2-Propenamide, N-hydroxy-3-[3-[(phenylamino)sulfonyl]phenyl]-, (2E)-](http://img.cochemist.com/ccimg/866400/866323-14-0.png)
![2-Propenamide, N-hydroxy-3-[3-[(phenylamino)sulfonyl]phenyl]-, (2E)-](http://img.cochemist.com/ccimg/866400/866323-14-0_b.png)
![2-Penten-4-ynamide,N-hydroxy-5-[3-[(phenylsulfonyl)amino]phenyl]-, (2E)-](http://img.cochemist.com/ccimg/151800/151720-43-3.png)
![2-Penten-4-ynamide,N-hydroxy-5-[3-[(phenylsulfonyl)amino]phenyl]-, (2E)-](http://img.cochemist.com/ccimg/151800/151720-43-3_b.png)
![Carbamic acid, [6-(hydroxyamino)-6-oxohexyl]-, 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/147500/147435-34-5.png)
![Carbamic acid, [6-(hydroxyamino)-6-oxohexyl]-, 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/147500/147435-34-5_b.png)
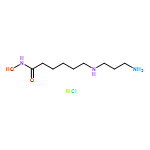
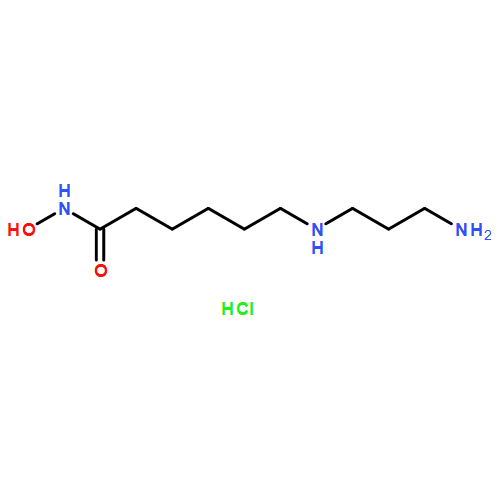



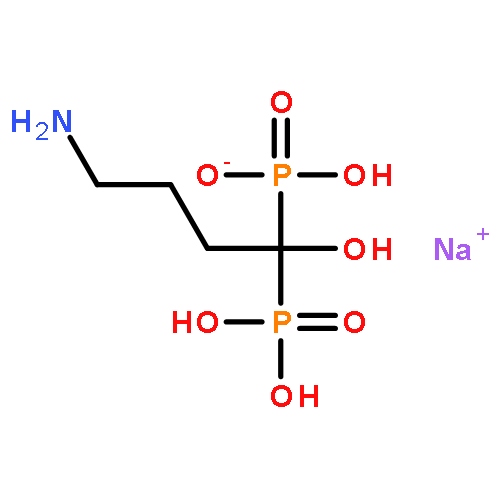


![(1S,5S)-1β,8-Dimethyl-4β-(1-methylethyl)spiro[4.5]deca-6,8-diene](http://img.cochemist.com/ccimg/90000/89955-08-8.png)
![(1S,5S)-1β,8-Dimethyl-4β-(1-methylethyl)spiro[4.5]deca-6,8-diene](http://img.cochemist.com/ccimg/90000/89955-08-8_b.png)
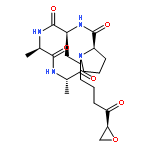


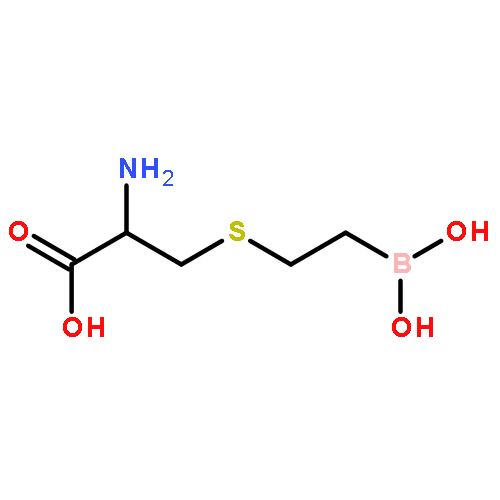
![L-Ornithine,N5-[(hydroxyamino)iminomethyl]-](http://img.cochemist.com/ccimg/53100/53054-07-2.png)
![L-Ornithine,N5-[(hydroxyamino)iminomethyl]-](http://img.cochemist.com/ccimg/53100/53054-07-2_b.png)


![Spiro[4.5]dec-7-ene, 1,8-dimethyl-4-(1-methylethenyl)-, (1R,4S,5R)-](http://img.cochemist.com/ccimg/28500/28477-64-7.png)
![Spiro[4.5]dec-7-ene, 1,8-dimethyl-4-(1-methylethenyl)-, (1R,4S,5R)-](http://img.cochemist.com/ccimg/28500/28477-64-7_b.png)









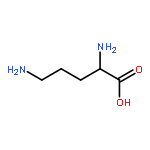
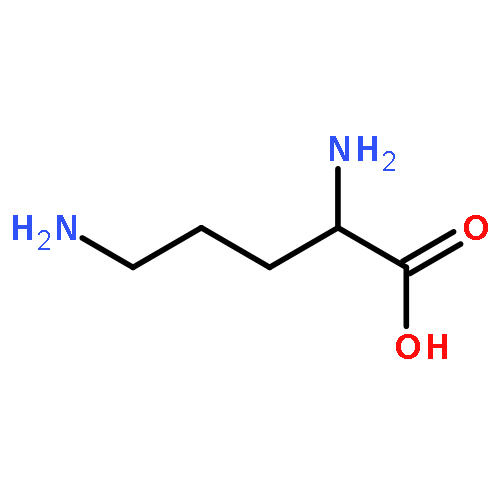
![Diphosphoric acid,P-[(2E,6E,10E)-3,7,11,15-tetramethyl-2,6,10,14-hexadecatetraen-1-yl] ester](http://img.cochemist.com/ccimg/6700/6699-20-3.png)
![Diphosphoric acid,P-[(2E,6E,10E)-3,7,11,15-tetramethyl-2,6,10,14-hexadecatetraen-1-yl] ester](http://img.cochemist.com/ccimg/6700/6699-20-3_b.png)