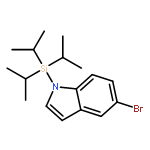
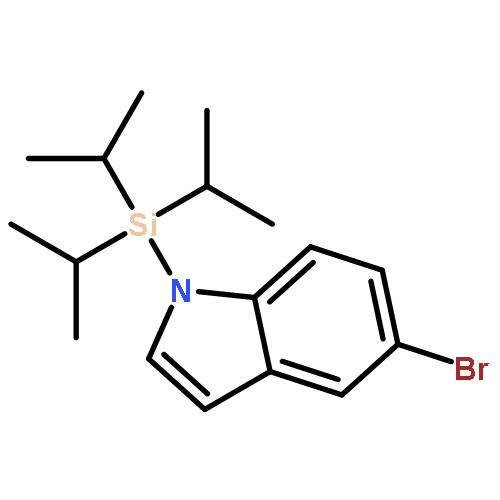
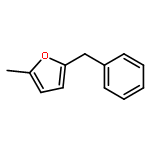
Co-reporter:Jiadi Zhang; Sheng-Chun Sha; Ana Bellomo; Nisalak Trongsiriwat; Feng Gao; Neil C. Tomson;Patrick J. Walsh
Journal of the American Chemical Society 2016 Volume 138(Issue 12) pp:4260-4266
Publication Date(Web):March 3, 2016
DOI:10.1021/jacs.6b01578
Metal-catalyzed carbon–carbon bond-forming reactions are a mainstay in the synthesis of pharmaceutical agents. A long-standing problem plaguing the field of transition metal catalyzed C–H functionalization chemistry is control of selectivity among inequivalent C–H bonds in organic reactants. Herein we advance an approach to direct site selectivity in the arylation of 2-benzylfurans founded on the idea that modulation of cooperativity in bimetallic catalysts can enable navigation of selectivity. The bimetallic catalysts introduced herein exert a high degree of control, leading to divergent site-selective arylation reactions of both sp2 and sp3 C–H bonds of 2-benzylfurans. It is proposed that the selectivity is governed by cation−π interactions, which can be modulated by choice of base and accompanying additives [MN(SiMe3)2, M = K or Li·12-crown-4].
Co-reporter:Ryan A. Zarkesh, Andrew S. Ichimura, Todd C. Monson, Neil C. Tomson and Mitchell R. Anstey
Dalton Transactions 2016 vol. 45(Issue 24) pp:9962-9969
Publication Date(Web):21 Mar 2016
DOI:10.1039/C6DT00422A
The redox-active bis(imino)acenapthene (BIAN) ligand was used to synthesize homoleptic aluminum, chromium, and gallium complexes of the general formula (BIAN)3M. The resulting compounds were characterized using X-ray crystallography, NMR, EPR, magnetic susceptibility and cyclic voltammetry measurements and modeled using both DFT and ab initio wavefunction calculations to compare the orbital contributions of main group elements and transition metals in ligand-based redox events. Complexes of this type have the potential to improve the energy density and electrolyte stability of grid-scale energy storage technologies, such as redox flow batteries, through thermodynamically-clustered redox events.
Co-reporter:Neil C. Tomson, Kamille D. Williams, Xuliang Dai, Stephen Sproules, Serena DeBeer, Timothy H. Warren and Karl Wieghardt
Chemical Science 2015 vol. 6(Issue 4) pp:2474-2487
Publication Date(Web):20 Feb 2015
DOI:10.1039/C4SC03294B
Three [Me2NN]Cu(η2-L2) complexes (Me2NN = HC[C(Me)NAr]2; L2 = PhNO (2), (3), PhCHCH2 (4); Ar = 2,6-Me2-C6H3; ArF = 3,5-(CF3)2-C6H3) have been studied by Cu K-edge X-ray absorption spectroscopy, as well as single- and multi-reference computational methods (DFT, TD-DFT, CASSCF, MRCI, and OVB). The study was extended to a range of both known and theoretical compounds bearing 2p-element donors as a means of deriving a consistent view of how the pre-edge transition energy responds in systems with significant ground state covalency. The ground state electronic structures of many of the compounds under investigation were found to be strongly influenced by correlation effects, resulting in ground state descriptions with majority contributions from a configuration comprised of a Cu(II) metal center anti-ferromagentically coupled to radical anion O2, PhNO, and ligands. In contrast, the styrene complex 4, which displays a Cu K pre-edge transition despite its formal d10 electron configuration, exhibits what can best be described as a Cu(I):(styrene)0 ground state with strong π-backbonding. The Cu K pre-edge features for these complexes increase in energy from 1 to 4, a trend that was tracked to the percent Cu(II)-character in the ground state. The unexpected shift to higher pre-edge transition energies with decreasing charge on copper (QCu) contributed to an assignment of the pre-edge features for these species as arising from metal-to-ligand charge transfer instead of the traditional Cu1s → Cu3d designation.
Co-reporter:Ryan A. Zarkesh, Andrew S. Ichimura, Todd C. Monson, Neil C. Tomson and Mitchell R. Anstey
Dalton Transactions 2016 - vol. 45(Issue 24) pp:NaN9969-9969
Publication Date(Web):2016/03/21
DOI:10.1039/C6DT00422A
The redox-active bis(imino)acenapthene (BIAN) ligand was used to synthesize homoleptic aluminum, chromium, and gallium complexes of the general formula (BIAN)3M. The resulting compounds were characterized using X-ray crystallography, NMR, EPR, magnetic susceptibility and cyclic voltammetry measurements and modeled using both DFT and ab initio wavefunction calculations to compare the orbital contributions of main group elements and transition metals in ligand-based redox events. Complexes of this type have the potential to improve the energy density and electrolyte stability of grid-scale energy storage technologies, such as redox flow batteries, through thermodynamically-clustered redox events.
Co-reporter:Neil C. Tomson, Kamille D. Williams, Xuliang Dai, Stephen Sproules, Serena DeBeer, Timothy H. Warren and Karl Wieghardt
Chemical Science (2010-Present) 2015 - vol. 6(Issue 4) pp:NaN2487-2487
Publication Date(Web):2015/02/20
DOI:10.1039/C4SC03294B
Three [Me2NN]Cu(η2-L2) complexes (Me2NN = HC[C(Me)NAr]2; L2 = PhNO (2), (3), PhCHCH2 (4); Ar = 2,6-Me2-C6H3; ArF = 3,5-(CF3)2-C6H3) have been studied by Cu K-edge X-ray absorption spectroscopy, as well as single- and multi-reference computational methods (DFT, TD-DFT, CASSCF, MRCI, and OVB). The study was extended to a range of both known and theoretical compounds bearing 2p-element donors as a means of deriving a consistent view of how the pre-edge transition energy responds in systems with significant ground state covalency. The ground state electronic structures of many of the compounds under investigation were found to be strongly influenced by correlation effects, resulting in ground state descriptions with majority contributions from a configuration comprised of a Cu(II) metal center anti-ferromagentically coupled to radical anion O2, PhNO, and ligands. In contrast, the styrene complex 4, which displays a Cu K pre-edge transition despite its formal d10 electron configuration, exhibits what can best be described as a Cu(I):(styrene)0 ground state with strong π-backbonding. The Cu K pre-edge features for these complexes increase in energy from 1 to 4, a trend that was tracked to the percent Cu(II)-character in the ground state. The unexpected shift to higher pre-edge transition energies with decreasing charge on copper (QCu) contributed to an assignment of the pre-edge features for these species as arising from metal-to-ligand charge transfer instead of the traditional Cu1s → Cu3d designation.
.jpg)