Co-reporter:Markus Scharfenberg, Jan Seiwert, Maximilian Scherger, Jasmin Preis, Moritz Susewind, and Holger Frey
Macromolecules September 12, 2017 Volume 50(Issue 17) pp:6577-6577
Publication Date(Web):August 21, 2017
DOI:10.1021/acs.macromol.7b01131
Multiarm star copolymers, consisting of hyperbranched poly(ethylene oxide) (hbPEO) or poly(butylene oxide) (hbPBO) polyether copolymers with glycerol branching points as a core, and linear aliphatic polycarbonate arms generated from carbon dioxide (CO2) and epoxide monomers, were synthesized via a “core-first” approach in two steps. First, hyperbranched polyether polyols were prepared by anionic copolymerization of ethylene oxide or 1,2-butylene oxide with 8–35% glycidol with molecular weights between 800 and 389,000 g·mol–1. Second, multiple arms were grown via immortal copolymerization of CO2 with propylene oxide or 1,2-butylene oxide using the polyether polyols as macroinitiators and (R,R)-(salcy)-CoCl as a catalyst in a solvent-free procedure. Molecular weights up to 812,000 g·mol–1 were obtained for the resulting multiarm polycarbonates, determined by online viscometry with universal calibration and 1H NMR. Comparing the synthesis of different multiarm star polycarbonates, a combination of a highly reactive macroinitiator with a less reactive epoxide monomer was found to be most suitable to obtain well-defined structures containing up to 88 mol% polycarbonate. The multiarm star copolymers were investigated with respect to their thermal properties, intrinsic viscosity, and potential application as polyols for polyurethane synthesis. Glass transition temperatures in the range from −41 to +25 °C were observed. The intrinsic viscosity could be adjusted between 5.4 and 17.3 cm3·g–1 by varying the ratio of polyether units and polycarbonate units.
Co-reporter:Jan Blankenburg, Manfred Wagner, and Holger Frey
Macromolecules November 28, 2017 Volume 50(Issue 22) pp:8885-8885
Publication Date(Web):November 9, 2017
DOI:10.1021/acs.macromol.7b01324
Multi-amino-functional poly(propylene oxide) (PPO) copolymers were synthesized by the anionic ring-opening copolymerization (AROP) of N,N-diethyl glycidyl amine (DEGA) and propylene oxide (PO). A solvent free synthesis route using potassium counterions and crown ether for the AROP enabled controlled (co)polymerization with full conversion. The strategy provided access to PPO-b-PDEGA block copolymers, statistical PPO-co-PDEGA copolymers, and, for the first time, PDEGA homopolymer. Molecular weights in the range of 1400 to 4200 g/mol (Mn) and dispersities (Mw/Mn) below 1.1 were obtained. Both the kinetics and resulting microstructure of the statistical copolymerization were investigated by in situ 1H NMR spectroscopy, revealing reactivity ratios of rPO = 1.7 and rDEGA = 0.9. The polyethers exhibit thermoresponsive behavior in aqueous solution, showing cloud points in the range 14–57 °C under neutral conditions. Because of the amine groups, the copolymer structures are strongly affected by the pH of the solution, and therefore the cloud points are highly pH-dependent. By changing the pH of the polymer solution, the cloud points can be tuned between 5 and 100 °C, as studied by turbidity measurements. The copolymers offer new potential for PPO-based structures for surface modification, biomedical purposes and pharmaceutical application.
Co-reporter:Benjamin Klöckner, Kerstin Niederer, Ana Fokina, Holger Frey, and Rudolf Zentel
Macromolecules May 23, 2017 Volume 50(Issue 10) pp:3779-3779
Publication Date(Web):May 12, 2017
DOI:10.1021/acs.macromol.7b00217
To combine several inorganic components with organic material in a controlled special and permanent manner still remains a difficult issue. Two specifically functionalized block copolymers were synthesized separately and combined in a second step. A heterofunctional poly(ethylene glycol) (PEG) block copolymer bearing a single amino unit, a short PEG spacer, and multiple catechol functionalities was obtained via anionic ring-opening polymerization (AROP). Using the reversible addition–fragmentation chain transfer (RAFT) radical polymerization technique, a semiconducting block copolymer with carbazole side groups was obtained. The second polyacrylate block contained reactive ester groups and was polymerized onto this hole conducting block. By substitution of the reactive esters with the amino functional PEG-catechol block copolymer and cysteamine methyl disulfide, a hole conducting polymer material with two orthogonal anchor groups for the coating of CdSe QDs, and also for TiO2, was obtained. TEM images show that upon coating of both materials we were able to obtain QDs homogeneously distributed at the surface of TiO2 nanoparticles. This spatial assembly is a consequence of the special directing features of the copolymer, possessing two orthogonal anchor groups combined in one material.
Co-reporter:Matthias Worm;Daniel Leibig;Carsten Dingels
ACS Macro Letters December 20, 2016 Volume 5(Issue 12) pp:1357-1363
Publication Date(Web):November 28, 2016
DOI:10.1021/acsmacrolett.6b00735
Polyethylene glycol (PEG) has been used for decades to improve the pharmacokinetic properties of protein drugs, and several PEG-protein conjugates are approved by the FDA. However, the nondegradability of PEG restricts its use to a limiting molecular weight to permit renal excretion. In this work, we introduce a simple strategy to overcome the nondegradability of PEG by incorporating multiple pH-sensitive vinyl ether moieties into the polyether backbone. Copolymerization of 3,4-epoxy-1-butene (EPB) with ethylene oxide via anionic ring-opening polymerization (AROP) provides access to allyl moieties that can be isomerized to pH-cleavable propenyl units (isoEPB). Well-defined P(EPB-co-EG) copolymers (Đ = 1.05–1.11) with EPB contents of ∼4 mol% were synthesized in a molecular weight range of 3000 to 10000 g mol–1. 1H NMR kinetic studies served to investigate acidic hydrolysis in a pH range of 4.4 to 5.4 and even allowed to distinguish between the hydrolysis rates of (E)- and (Z)-isoEPB units, demonstrating faster hydrolysis of the (Z)-isomer. SEC analysis of degradation products revealed moderate dispersities Đ of 1.6 to 1.8 and consistent average molecular weights Mn of ∼1000 g mol–1. The presence of a defined hydroxyl end group permits attachment to other functional molecules. The novel pH-degradable PEGs combine various desirable properties such as excellent long-term storage stability and cleavage in a physiologically relevant pH-range that render them promising candidates for biomedical application.
Co-reporter:Hannah Pohlit, Matthias Worm, Jens Langhanki, Elena Berger-Nicoletti, Till Opatz, and Holger Frey
Macromolecules December 12, 2017 Volume 50(Issue 23) pp:9196-9196
Publication Date(Web):November 29, 2017
DOI:10.1021/acs.macromol.7b01787
Heterobifunctional poly(ethylene glycol)s (PEGs) are key structures for bioconjugation in the context of the “PEGylation” strategy to enhance blood circulation times of, for example, peptide drugs or “stealth” liposomes. The formation of heterobifunctional PEGs from symmetric PEG diols is challenging because of limited yields of the targeted monofunctional product and difficulties associated with separation steps. On the basis of a detailed comparison of reaction conditions, we have investigated a “polymer desymmetrization” strategy to maximize the yields of monofunctional PEG tosylate. The tosylation reaction in the presence of the heterogeneous catalyst silver oxide and potassium iodide in a specific stoichiometric ratio proved to be highly efficient, resulting in 71–76% yield of monofunctional PEG depending on molecular weight, exceeding the expected value of 50% in a statistical reaction without addition of a catalyst. For characterization as well as for the preparative separation of monotosylated PEG, we developed a HPLC method, using an evaporative light scattering detector, enabling both analytical and semipreparative separation of monotosylated PEGs on gram scale up to 20 000 g mol–1. To demonstrate the efficiency of the procedure, an α-azido-ω-methacryloyl-PEG was prepared as a building block suitable for azide–alkyne click-type reactions that can be incorporated into polymer networks via radical polymerization. We click-functionalized α-azido-ω-methacryloyl-PEG with a mannose-functionalized alkyne to enable functionalization of nanogels for enhanced cellular uptake via the mannose receptor. The synthesis strategy is suitable for a broad range of applications in the field of PEGylation and for hydrogel and nanogel functionalization.
Co-reporter:Markus Scharfenberg, Silja Hofmann, Jasmin Preis, Jeannette Hilf, and Holger Frey
Macromolecules August 22, 2017 Volume 50(Issue 16) pp:6088-6088
Publication Date(Web):August 1, 2017
DOI:10.1021/acs.macromol.7b01276
Hyperbranched, multifunctional polycarbonate polyols based on CO2, cyclohexene oxide (CHO), and the “inimer” (initiator–monomer) (4-hydroxymethyl)cyclohexene oxide (HCHO) were prepared in one-pot syntheses. The related linear poly(hydroxymethyl cyclohexene carbonate) structures based on protected HCHO and postpolymerization deprotection were also synthesized as model compounds. The content of hydroxyl functionalities was adjustable for both linear and hyperbranched terpolymer systems. All CO2/epoxide polymerizations were catalyzed by the (R,R)-(salcy)-Co(III)Cl complex. The polycarbonates obtained were comprehensively investigated using various 1D and 2D NMR techniques, SEC, FT-IR, UV–vis spectroscopy, and contact angle measurements. Rigid polyols with molecular weights between 3600 and 9200 g mol–1 and moderate dispersity between 1.18 and 1.64 (Mw/Mn) were obtained. In addition, the materials were examined with respect to their thermal properties, intrinsic viscosity, and their three-dimensional structure. Glass transition temperatures in the range of 113–141 °C (linear) and 72–105 °C (hyperbranched) were observed. The intrinsic viscosity of the hyperbranched systems is in the range of 5.69–11.51 cm3 g–1 and mirrors their compact structure. The hyperbranched polyols were also studied regarding their successful reaction with phenyl isocyanate to convert the free hydroxyl groups into urethanes.
Co-reporter:Christian Schubert, Philip Dreier, Thong Nguyen, Kamil Maciol, Jan Blankenburg, Christian Friedrich, Holger Frey
Polymer 2017 Volume 121(Volume 121) pp:
Publication Date(Web):14 July 2017
DOI:10.1016/j.polymer.2017.05.030
•Synthesis of linear random polyglycerol copolymers.•Adjustable hydroxyl functionality.•Detailed rheological study.We introduce a two-step strategy for the synthesis of linear polyglycerols (linPG-OHx/OMey) with an adjustable degree of methylation (y=DM100). Ethoxy ethyl glycidyl ether (EEGE) and glycidyl methyl ether (GME) were copolymerized via the “activated monomer” polymerization technique, using tetraoctylammonium bromide (NOct4Br) as an initiator and triisobutylaluminum (i-Bu3Al) as a catalyst under mild conditions. Subsequent acidic cleavage of the acetal protective groups generates linear polyglycerols with different degree of methylation (DM) by varying the GME comonomer content between 10 and 91%. Size exclusion chromatography (SEC) evidenced good control over molecular weight and narrow to moderate polydispersity (PDI = 1.2–1.8). 1H NMR spectroscopy confirmed ideally random copolymerization of EEGE and GME (in situ 1H-NMR kinetics) and provided perfect agreement of the comonomer content with the targeted values. Thermoresponsive behavior in solution and lowering of cloud points with increasing degree of methylation was observed. Furthermore, the differently methylated polyglycerols were investigated with respect to their rheological properties in the melt. Comparison with the fully hydroxylated and permethylated polyglycerol provides new insights into the dynamic behavior of functional polyethers. A tremendous influence of DM on zero-shear viscosity and differences of up to 5 decades were observable at the same reference temperature (273 K). The trend of glass transition temperature and zero-shear viscosity in dependency of degree of methylation was described by mixing rules. To understand the changes in zero-shear viscosity, the “sticky” Rouse model was applied and led to an estimated association lifetime of stickers, i.e. hydroxyl groups of τs=4.9±1.8μs.Download high-res image (131KB)Download full-size image
Co-reporter:Jana Herzberger;Daniel Leibig;Jens Langhanki;Christian Moers;Till Opatz
Polymer Chemistry (2010-Present) 2017 vol. 8(Issue 12) pp:1882-1887
Publication Date(Web):2017/03/21
DOI:10.1039/C7PY00173H
A straight forward synthesis of poly(ethylene glycol) (PEG) with multiple alkyne groups distributed along the polymer chain is introduced. Direct access to clickable PEG is achieved by the monomer-activated anionic ring-opening copolymerization (AROP) of ethylene oxide (EO) with glycidyl propargyl ether (GPgE). Notably for successful polymerization no protection of the alkyne unit is required owing to the mild reaction conditions. Defined PEG-co-PGPgE and PGPgE (co)polymers with PDIs of 1.18–1.60 and molecular weights of Mn = 3000–9500 g mol−1 were prepared. In situ1H NMR kinetic studies revealed remarkably disparate reactivity ratios of rEO = 14.8 and rGPgE = 0.076, representing a pronounced compositional drift with EO rich segments close to the initiator and GPgE units near the chain terminus, i.e. copolymers with a steep monomer gradient. Copper(I)-catalyzed azide–alkyne cycloaddition (CuAAC) with mannopyranosyl azide leads to PEG-based glycopolymers and highlights the potential of alkyne-functional PEGs as a universal scaffold for a range of biomedical applications.
Co-reporter:Jan Seiwert;Jana Herzberger;Daniel Leibig
Macromolecular Rapid Communications 2017 Volume 38(Issue 1) pp:
Publication Date(Web):2017/01/01
DOI:10.1002/marc.201600457
The synthesis of thioether-bearing hyperbranched polyether polyols based on an AB/AB2 type copolymerization (cyclic latent monomers) is introduced. The polymers are prepared by anionic ring-opening multibranching copolymerization of glycidol and 2-(methylthio)ethyl glycidyl ether (MTEGE), which is conveniently accessible in a single etherification step. Slow monomer addition provides control over molecular weights. Moderate dispersities (Đ = 1.48–1.85) are obtained, given the hyperbranched structure. In situ 1H NMR copolymerization kinetics reveal reactivity ratios of rG = 3.7 and rMTEGE = 0.27. Using slow monomer addition, copolymer composition can be systematically varied, allowing for the adjustment of the hydroxyl/thioether ratio, the degree of branching (DB = 0.36–0.48), thermal properties, and cloud point temperatures in aqueous solution in the range of 29–75 °C. Thioether oxidation to sulfoxides enables to tailor the copolymers' solubility profile. Use of these copolymers as a versatile, multifunctional platform for orthogonal modification is highlighted. The methyl sulfide groups can be selectively alkoxylated, using propylene oxide, allyl glycidyl ether, or furfuryl glycidyl ether, resulting in functional hyperbranched polyelectrolytes. Reaction of the alcohol groups with benzyl isocyanate demonstrates successful orthogonal functionalization.
Co-reporter:Jana Herzberger, Kerstin Niederer, Hannah Pohlit, Jan Seiwert, Matthias Worm, Frederik R. Wurm, and Holger Frey
Chemical Reviews 2016 Volume 116(Issue 4) pp:2170
Publication Date(Web):December 29, 2015
DOI:10.1021/acs.chemrev.5b00441
The review summarizes current trends and developments in the polymerization of alkylene oxides in the last two decades since 1995, with a particular focus on the most important epoxide monomers ethylene oxide (EO), propylene oxide (PO), and butylene oxide (BO). Classical synthetic pathways, i.e., anionic polymerization, coordination polymerization, and cationic polymerization of epoxides (oxiranes), are briefly reviewed. The main focus of the review lies on more recent and in some cases metal-free methods for epoxide polymerization, i.e., the activated monomer strategy, the use of organocatalysts, such as N-heterocyclic carbenes (NHCs) and N-heterocyclic olefins (NHOs) as well as phosphazene bases. In addition, the commercially relevant double-metal cyanide (DMC) catalyst systems are discussed. Besides the synthetic progress, new types of multifunctional linear PEG (mf-PEG) and PPO structures accessible by copolymerization of EO or PO with functional epoxide comonomers are presented as well as complex branched, hyperbranched, and dendrimer like polyethers. Amphiphilic block copolymers based on PEO and PPO (Poloxamers and Pluronics) and advances in the area of PEGylation as the most important bioconjugation strategy are also summarized. With the ever growing toolbox for epoxide polymerization, a “polyether universe” may be envisaged that in its structural diversity parallels the immense variety of structural options available for polymers based on vinyl monomers with a purely carbon-based backbone.
Co-reporter:Jana Herzberger; Karl Fischer; Daniel Leibig; Matthias Bros; Raphael Thiermann
Journal of the American Chemical Society 2016 Volume 138(Issue 29) pp:9212-9223
Publication Date(Web):July 2, 2016
DOI:10.1021/jacs.6b04548
Poly(ethylene glycol) (PEG) is a widely used biocompatible polymer. We describe a novel epoxide monomer with methyl-thioether moiety, 2-(methylthio)ethyl glycidyl ether (MTEGE), which enables the synthesis of well-defined thioether-functional poly(ethylene glycol). Random and block mPEG-b-PMTEGE copolymers (Mw/Mn = 1.05–1.17) were obtained via anionic ring opening polymerization (AROP) with molecular weights ranging from 5 600 to 12 000 g·mol–1. The statistical copolymerization of MTEGE with ethylene oxide results in a random microstructure (rEO = 0.92 ± 0.02 and rMTEG E = 1.06 ± 0.02), which was confirmed by in situ 1H NMR kinetic studies. The random copolymers are thermoresponsive in aqueous solution, with a wide range of tunable transition temperatures of 88 to 28 °C. In contrast, mPEG-b-PMTEGE block copolymers formed well-defined micelles (Rh ≈ 9–15 nm) in water, studied by detailed light scattering (DLS and SLS). Intriguingly, the thioether moieties of MTEGE can be selectively oxidized into sulfoxide units, leading to full disassembly of the micelles, as confirmed by detection of pure unimers (DLS and SLS). Oxidation-responsive release of encapsulated Nile Red demonstrates the potential of these micelles as redox-responsive nanocarriers. MTT assays showed only minor effects of the thioethers and their oxidized derivatives on the cellular metabolism of WEHI-164 and HEK-293T cell lines (1–1000 μg·mL–1). Further, sulfonium PEG polyelectrolytes can be obtained via alkylation or alkoxylation of MTEGE, providing access to a large variety of functional groups at the charged sulfur atom.
Co-reporter:Jana Herzberger, Daniel Leibig, Johannes C. Liermann, and Holger Frey
ACS Macro Letters 2016 Volume 5(Issue 11) pp:1206
Publication Date(Web):October 13, 2016
DOI:10.1021/acsmacrolett.6b00701
Detailed understanding of the monomer distribution in copolymers is essential to tailor their properties. For the first time, we have been able to utilize in situ 1H NMR spectroscopy to monitor the monomer-activated anionic ring opening copolymerization (AROP) of ethylene oxide (EO) with a glycidyl ether comonomer, namely, ethoxy ethyl glycidyl ether (EEGE). We determine reactivity ratios and draw a direct comparison to conventional oxyanionic ROP. Surprisingly, the respective monomer reactivities differ strongly between the different types of AROP. Under conventional oxyanionic conditions similar monomer reactivities of EO and EEGE are observed, leading to random structures (rEO = 1.05 ± 0.02, rEEGE = 0.94 ± 0.02). Addition of a cation complexing agent (18-crown-6) showed no influence on the relative reactivity of EO and EEGE (rEO = rEEGE = 1.00 ± 0.02). In striking contrast, monomer-activated AROP produces very different monomer reactivities, affording strongly tapered copolymer structures (rEO = 8.00 ± 0.16, rEEGE = 0.125 ± 0.003). These results highlight the importance of understanding reactivity ratios of comonomer pairs under certain polymerization conditions, at the same time demonstrating the ability to generate both random and strongly tapered P(EO-co-EEGE) polyethers by simple one-pot statistical anionic copolymerization. These observations may be generally valid for the copolymerization of EO and glycidyl ethers.
Co-reporter:Matthias Worm;Biao Kang;Carsten Dingels;Frederik R. Wurm
Macromolecular Rapid Communications 2016 Volume 37( Issue 9) pp:775-780
Publication Date(Web):
DOI:10.1002/marc.201600080
Co-reporter:Daniel Leibig, Axel H. E. Müller, and Holger Frey
Macromolecules 2016 Volume 49(Issue 13) pp:4792-4801
Publication Date(Web):June 29, 2016
DOI:10.1021/acs.macromol.6b00831
The catechol-containing vinyl monomers 4-vinylcatechol acetonide (4-VCA) and 3-vinylcatechol acetonide (3-VCA) are introduced for carbanionic polymerization in THF, using sec-butyllithium as an initiator. Molecular weights (Mn) ranging from 3000 to 80 000 g mol–1 were obtained, with polydispersities (Mw/Mn) below 1.10 for 4-VCA and 1.15 for 3-VCA homopolymerization. Furthermore, block copolymers and gradient copolymers with styrene have been prepared via living carbanionic copolymerization. The reactivity of the new monomers 4-VCA and 3-VCA in the copolymerization with styrene and the resulting monomer gradient in the copolymer chains were investigated via in situ 1H NMR spectroscopic kinetic studies in toluene-d8. The results show lower reactivity of the 4-VCA monomer than styrene (rS = 4.0, r4-VCA = 0.24) and a higher reactivity than styrene for 3-VCA (r3-VCA = 2.4, rS = 0.48). Well-defined copolymers of styrene and 4-VCA exhibit a strong gradient structure within the polymer chains with the catechol functionalities preferentially incorporated near the chain terminus. However, in the case of 3-VCA, the gradient structure of the copolymers is reversed, and the catechol functionalities are preferentially incorporated in the vicinity of the initiator. The direction of the monomer gradient in the copolymers can be predicted from the difference of the chemical shift of the β-carbon signal of the respective vinyl monomers in 13C NMR spectra, which has general implications for the copolymerization of vinyl monomers. All polymers were characterized by 1H NMR spectroscopy, size exclusion chromatography (SEC), MALDI-ToF mass spectrometry, and differential scanning calorimetry (DSC). Quantitative cleavage of the acetonide protecting group under mild acidic conditions rendered well-defined poly(vinyl catechol)s, which were used for pH-sensitive precipitation of iron(III) cations and for surface coating on a variety of materials, showing very stable and permanent catechol-promoted adhesion.
Co-reporter:Daniel Leibig, Jan Seiwert, Johannes C. Liermann, and Holger Frey
Macromolecules 2016 Volume 49(Issue 20) pp:7767-7776
Publication Date(Web):October 4, 2016
DOI:10.1021/acs.macromol.6b01477
Copolymerization of established epoxide monomers with glycidol (G) is a key reaction to prepare branched or hyperbranched polyethers. The kinetics of the multibranching anionic ring-opening copolymerization of glycidol (a cyclic latent AB2 monomer) with ethylene oxide (EO), propylene oxide (PO), and 1,2-butylene oxide (BO; cyclic latent AB monomers), respectively, in dimethyl sulfoxide was studied. Online 1H NMR spectroscopy was employed for in situ monitoring of the individual monomer consumption during the entire course of the statistical copolymerization. Varying the counterion, both the cesium alkoxide and potassium alkoxide initiated copolymerization were studied and compared. From the individual monomer consumption, reactivity ratios were calculated. The reactivity ratio of the alkylene oxides decreases from 0.44 to 0.11 with increasing alkyl chain length on going from EO to BO. Unexpectedly, glycidol was found to exhibit a higher reactivity ratio in each copolymerization, with reactivity ratios ranging from 2.34 (with EO) to 7.94 (copolymerization with BO). Different counterions had an impact on absolute reaction rates, however, relative monomer reactivities remained unchanged. The reactivity ratios determine both the molecular weight distribution and the topology as well as the degree of branching (DB) of the respective branched copolymers, implying a change from a hyperbranched random copolymer (glycidol/EO) to a multiarm star structure with increasing side chain length of the alkylene comonomer.
Co-reporter:Christian Schubert, Carina Osterwinter, Christoph Tonhauser, Martina Schömer, Daniel Wilms, Holger Frey, and Christian Friedrich
Macromolecules 2016 Volume 49(Issue 22) pp:8722-8737
Publication Date(Web):November 4, 2016
DOI:10.1021/acs.macromol.6b00674
Melt rheology and thermal phase transition of a series of hyperbranched polyglycerol samples (hbPG) (DB ≈ 60%) in a broad molecular weight range (Mn = 600–440 000 g/mol) were investigated and correlated to both molecular weight and nature of the end group (hydroxyl vs permethylated and trimethylsilylated). The well-characterized and defined flexible polyethers are particularly suitable to shed light on the linear viscoelastic behavior with respect to (i) hyperbranched topology and (ii) hydrogen bond interactions, particularly in comparison to the perfectly linear polyglycerol counterparts studied recently [Osterwinter, C.; Macromolecules 2015, 48, 119−130]. We present a detailed examination of differences found in the characteristic moduli as a consequence of functionality and topology leading to an estimation of both a stickiness parameter and a connectivity parameter of hyperbranched molecules. The appearance of a plateau region of the dynamic moduli indicates entanglement behavior, although the determined apparent entanglement molecular weight Me ≈ 6000 g/mol is significantly higher than the molecular weight between two branching points (Mx ≪ Me). Zero shear viscosities and terminal relaxation times show a unique scaling behavior with respect to the nature of the multiple end groups of the hyperbranched topology, suggesting entanglement transitions with a critical molecular weight around 55 000 g/mol. Most striking and in pronounced contrast to linear PG as the perfect linear analogue, the derived structure–property relationships depend on the functionality of the repeating unit of the polymer.
Co-reporter:Eva-Maria Christ, Jana Herzberger, Mirko Montigny, Wolfgang Tremel, and Holger Frey
Macromolecules 2016 Volume 49(Issue 10) pp:3681-3695
Publication Date(Web):May 6, 2016
DOI:10.1021/acs.macromol.6b00113
Cyano-functional polyether copolymers based on THF were prepared via cationic ring-opening copolymerization of THF with cyano ethylene oxide (CEO). The CEO content of poly(tetrahydrofuran) (polyTHF) based copolymers varied from 3.3 to 29.3%, and molecular weights ranged from 5100 to 31900 g·mol–1 with Mw/Mn in the range of 1.31 to 1.74 (SEC in THF, PS standards). The polymerization was conducted with methyl trifluoromethanesulfonate (MeOTf) as an initiator. Kinetic studies concerning incorporation of both monomers were performed via NMR spectroscopy. The cyano groups at the poly(THF-co-CEO) copolymers enable direct access to amino (polyTHF–NH2) and carboxyl groups (polyTHF–COOH) in facile one-step procedures, respectively. The modified copolymers were characterized via NMR, MALDI–ToF mass, and FT-IR spectroscopy. Thermal properties of the materials were studied via differential scanning calorimetry (DSC), demonstrating a gradual decrease of the melting points with increasing amount of CEO in the copolymers (from 30 °C for 3.3% CEO to 21 °C for 8.4% CEO). After postmodification to carboxylic acid groups the melting points decrease from 26 to 18 °C in the series of copolymers. Contact angles of water on thin films of the polymers can be tuned in a wide range from 72.7° to 17.8° by varying the CEO fraction as well as by postmodification. Crystallization studies of CaCO3 with water-soluble polyTHF–COOH revealed the composition-dependent inhibition of calcite growth, with crystallite size in the mineralization process being controlled by the amount of carboxylic acid groups at the poly(THF) copolymers.
Co-reporter:Kerstin Niederer, Christoph Schüll, Daniel Leibig, Tobias Johann, and Holger Frey
Macromolecules 2016 Volume 49(Issue 5) pp:1655-1665
Publication Date(Web):February 19, 2016
DOI:10.1021/acs.macromol.5b02441
A protected catechol-containing epoxide monomer, catechol acetonide glycidyl ether (CAGE), is introduced. CAGE is conveniently obtained in three steps and enables the incorporation of surface-active catechol moieties into a broad variety of hydrophilic and biocompatible polyether architectures by copolymerization. Via acidic cleavage of the acetal protecting groups, the polymer-attached catechol functionalities are liberated and available for surface attachment or metal complexation. CAGE has been copolymerized with ethylene oxide and glycidol to obtain both linear poly(ethylene glycol) and hyperbranched polyglycerol copolymers, respectively, with multiple surface-adhesive catechol moieties. The CAGE content in the copolymers was varied from 1 to 16%, and all polymers exhibit moderate polydispersity (linear: Mw/Mn = 1.05–1.33; hyperbranched: Mw/Mn = 1.44–1.86). In situ kinetic studies of the simultaneous copolymerization of EO and CAGE via NMR spectroscopy have been performed to determine the microstructure of the linear poly(ethylene oxide-co-catechol acetonide glycidyl ether), P(EO-co-CAGE), copolymers. EO shows slightly higher reactivity than CAGE (rEO = 1.14, rCAGE = 0.88), leading to an almost ideally random copolymerization. Because of the catechol units, the copolymers form pH-induced cross-linked networks through metal–ligand interactions. ABA triblock copolymers of the type PCAGE-b-PEG-b-PCAGE formed highly swellable hydrogels upon addition of FeCl3. Furthermore, static water contact angle measurements demonstrate an increase in the hydrophilicity of iron, PTFE, and PVC surfaces after coating with catechol-functional mf-PEGs.
Co-reporter:Jan Seiwert, Daniel Leibig, Ulrike Kemmer-Jonas, Marius Bauer, Igor Perevyazko, Jasmin Preis, and Holger Frey
Macromolecules 2016 Volume 49(Issue 1) pp:38-47
Publication Date(Web):December 23, 2015
DOI:10.1021/acs.macromol.5b02402
Hyperbranched poly(butylene oxide) polyols have been synthesized by multibranching anionic ring-opening copolymerization of 1,2-butylene oxide and glycidol. Systematic variation of the composition from 24 to 74% glycidol content resulted in a series of moderately distributed copolymers (Đ = 1.41–1.65, SEC), albeit with limited molecular weights in the solvent-free batch process in the range of 900–1300 g mol–1 (apparent Mn determined by SEC with PEG standards). In situ monitoring of the copolymerization kinetics by 1H NMR showed a pronounced compositional drift with respect to the monomer feed, indicating a strongly tapered microstructure caused by the higher reactivity of glycidol. In the case of slow monomer addition considerably higher apparent molecular weights up to 8500 g mol–1 were obtained (SEC). By alteration of the comonomer ratio, aqueous solubility of the hyperbranched copolymers could be tailored, resulting in well-defined cloud points between 20 and 84 °C. Glass transition temperatures between −60 and −29 °C were observed for the resulting polyether polyols. High degrees of branching (DB) between 0.45 and 0.77 were calculated from inverse gated (IG) 13C NMR. Online viscosimetry and analytical ultracentrifugation (AUC) were employed to study hydrodynamic properties and to establish a universal calibration curve for the determination of absolute molecular weights. This resulted in Mw values between 2100 and 35 000 g mol–1 that were generally 2–3 times higher than the apparent values determined by SEC with linear PEG standards.
Co-reporter:Jan Morsbach, Axel H. E. Müller, Elena Berger-Nicoletti, and Holger Frey
Macromolecules 2016 Volume 49(Issue 14) pp:5043-5050
Publication Date(Web):July 12, 2016
DOI:10.1021/acs.macromol.6b00975
Aiming at systematic variation of the parameter dispersity, Đ (or “polydispersity”), living polymers with predictable dispersity (Đ = 1.15–2.20) and controlled molecular weights (Mn = 3200–18 500 g mol–1) were prepared via carbanionic polymerization. The approach relies on a continuous flow reactor equipped with a tangential four-way jet micromixing device. By varying the total flow rate, the mixing efficiency of the initiator (sec-BuLi) and the corresponding vinyl monomers is controlled, resulting in polymers with predefined dispersity, while the number-average molecular weight, Mn, is kept constant. In this manner living polystyrene (PS), poly(p-methylstyrene) (PpMeS), and poly(2-vinylpyridine) (P2VP) samples with systematically varied Đ (studied by SEC) were prepared. All polymerizations were carried out at room temperature in a 50:50 solvent mixture of THF with either hexane for PS and PpMeS or with benzene for the polymerization of 2VP. To prove the living character of all polymer chains of the distributions obtained, all carbanionic chains were labeled, i.e., end functionalized via addition of an epoxide (benzyl glycidyl ether, BGE) as a termination reagent, when full conversion of the monomer was reached. Subsequent MALDI-ToF characterization confirmed the living character of all chains of the distributions. This is key for the further generation of complex polymer architectures with tailored polydispersity. Using the living carbanions with different dispersity, in an exploratory study, PS-b-PI block copolymers with controlled dispersity of the styrene block have been prepared via direct addition of isoprene as a second monomer.
Co-reporter:Jan Morsbach, Johannes Elbert, Christian Rüttiger, Svenja Winzen, Holger Frey, and Markus Gallei
Macromolecules 2016 Volume 49(Issue 9) pp:3406-3414
Publication Date(Web):April 21, 2016
DOI:10.1021/acs.macromol.6b00514
The synthesis of high-molecular-weight, well-defined poly(vinylferrocene)-block-poly(ethylene glycol) (PVFc-b-PEG) diblock copolymers (Mn = 13 000–44 000 g mol–1; Đ = 1.29–1.34) with precisely one allyl group at the junction point is introduced. Allyl glycidyl ether (AGE) was used to end-functionalize PVFc, resulting in hydroxyl functional macroinitiators for the oxyanionic polymerization of ethylene oxide. The self-assembly behavior of the amphiphilic PVFc-b-PEG copolymers in water has been investigated in a detailed manner, using dynamic light scattering (DLS) and transmission electron microscopy (TEM). The redox activity of the PVFc block was confirmed by UV/vis spectroscopy, while cyclovoltammetry (CV) measurements were carried out to support the stability and full reversibility of the ferrocene/ferrocenium redox couple. Both formation and dissociation of the macromolecular self-assemblies in aqueous solution via oxidation and reduction of the PVFc segments were evidenced by TEM and DLS. The dye Nile Red was used as model compound to investigate the stabilization of a water-insoluble molecule in aqueous solution by the block copolymers via encapsulation inside micellar structures. Oxidation of the PVFc segments lead to instantaneous and quantitative release of the dye. Furthermore, incorporation of the allyl moiety at the block junction point was used to cross-link the shell of the compartments. By this strategy a stable incorporation of the dye was achieved while triggered release via oxidation led to quantitative liberation.
Co-reporter:Jeannette Hilf;Markus Scharfenberg;Jeffrey Poon;Christian Moers
Macromolecular Rapid Communications 2015 Volume 36( Issue 2) pp:174-179
Publication Date(Web):
DOI:10.1002/marc.201400504
Co-reporter:Rebecca Klein;Fabian Übel
Macromolecular Rapid Communications 2015 Volume 36( Issue 20) pp:1822-1828
Publication Date(Web):
DOI:10.1002/marc.201500400
Co-reporter:Anna M. Fischer, Christoph Schüll, Holger Frey
Polymer 2015 Volume 72() pp:436-446
Publication Date(Web):18 August 2015
DOI:10.1016/j.polymer.2015.04.047
Sn(Oct)2-catalyzed synthesis of hyperbranched poly(glycolide) copolymers with glycerol branching points in the backbone is possible via ring-opening multi-branching copolymerization (ROMBP) of glycolide and 5HDON (5-hydroxymethyl-1,4-dioxan-2-one). Using this strategy, well-defined and soluble branched polyesters with apparent molecular weights (Mn) in the range of 1300–2000 g mol−1 and varying comonomer content (5HDON/glycolide = 30:70–70:30) were obtained. 2D NMR spectroscopy, thermal analysis and MALDI-TOF mass spectrometry confirmed the successful incorporation of both monomers and the resulting branched structure. Multiple end group functionality offers the possibility for further post-polymerization modification, rendering the materials interesting with respect to processing of PGA. Potential applications range from novel polyurethanes to biomedical purposes.
Co-reporter:Yaming Yu and Holger Frey
Langmuir 2015 Volume 31(Issue 48) pp:13101-13106
Publication Date(Web):November 12, 2015
DOI:10.1021/acs.langmuir.5b03243
Antifouling thin films derived from charged hyperbranched polyglycerol (hbPG) layers were fabricated and evaluated. The anionic hbPG (a-hbPG) monolayers and cationic hbPG/anionic hbPG (c/a-hbPG) bilayers were adsorbed on the underlying self-assembled monolayers (SAMs) of cysteamine and 3-mercaptopropionic acid (3-MPA) by electrostatic interaction, respectively, and their procession was monitored by surface plasmon resonance spectroscopy (SPR). The adsorption of bovine serum albumin (BSA) and fibrinogen on the premade a-hbPG and c/a-hbPG thin films was measured and the capability of these thin films to resist nonspecific protein adsorption was evaluated by SPR as well. It is observed that the c/a-hbPG bilayer films possessed good antifouling properties. With c/a-hbPG bilayers consisting of higher molecular weight a-hbPG, the adsorption of BSA and fibrinogen were as low as 0.015 ng/mm–2 and 0.0076 ng/mm–2, respectively, comparable to the traditionally ultralow antifouling surfaces (<0.05 ng/mm–2 of nonspecific protein adsorption). This work proved that the charged hbPG thin films can strongly reduce the nonspecific protein adsorption and have the promise for the antifouling coatings with improved performance.
Co-reporter:Carina Osterwinter, Christian Schubert, Christoph Tonhauser, Daniel Wilms, Holger Frey, and Christian Friedrich
Macromolecules 2015 Volume 48(Issue 1) pp:119-130
Publication Date(Web):December 18, 2014
DOI:10.1021/ma501674x
Viscoelastic properties of linear, hydroxyl-functional polymers are only little understood with respect to the effect of functional group interactions. Melt rheology and thermal phase transitions of linear polyethers (polyglycerol, linPG-OH) and their methylated analogues (linPG-OMe) in a broad molecular weight range (Mn = 1–100 kg/mol) with low polydispersities (PDI) have been investigated as a general model for hydroxyl-functional polymers with respect to their functionality and hydrogen bond interactions. We provide detailed insight into the rheodynamics of nonentangled and well-entangled polyethers bearing one functional group per monomer unit. Booij–Palmen plots (BBP) revealed failure of the time–temperature superposition principle (TTS) for both types of polymers in the segmental relaxation region, while TTS holds in the terminal relaxation region. The characteristic modulus of linPG-OMe derived from the BBP clearly reflects the transition from the nonentangled to the fully entangled state with increasing molecular weight. Quantitative analysis of these data allows for different estimates of the entanglement molecular weight, which is approximately 14 kg/mol. In case of linPG-OH a lower apparent entanglement molecular weight (8 kg/mol) leads to estimated 36 entanglement interactions in a cube of 10 nm edge length together with 47 association sites in the same volume. This can be determined from the molecular-weight-independent plateau modulus only, which is significantly lower than for linPG-OMe. This is explained as a consequence of the overlay of an entanglement network and an association network created by hydrogen bonding of the OH groups with themselves and with the ether linkages.
Co-reporter:Igor Perevyazko, Jan Seiwert, Martina Schömer, Holger Frey, Ulrich S. Schubert, and Georges M. Pavlov
Macromolecules 2015 Volume 48(Issue 16) pp:5887-5898
Publication Date(Web):August 13, 2015
DOI:10.1021/acs.macromol.5b01020
Hyperbranched poly(ethylene glycol) copolymers were synthesized by random anionic ring-opening multibranching copolymerization of ethylene oxide with glycidol as a branching agent, leading to poly(ethylene glycol) structure with glycerol branching points. Extending the available range of molar masses by novel synthesis strategies, a limited extent of control over the degree of polymerization was achieved by variation of the solvent in this copolymerization. Generally, absolute molar mass characterization of hyperbranched polymers still represents an unresolved challenge. A series of the hyperbranched poly(ethylene glycol)-co-(glycerol) copolymers (hbPEGs) of a wide range of molar masses (1400 < M < 1 700 000 g mol–1), degree of branching (DB) = 0.04–0.54, and moderate polydispersity (Mw/Mn) ≈ 2.1 ± 0.2 were studied, in both water and dimethylformamide by the methods of molecular hydrodynamics. Analytical ultracentrifugation, intrinsic viscosity, translational diffusion measurements, and SEC were combined. Molar masses of hbPEGs were estimated from the comparison of the velocity sedimentation and translational diffusion coefficients, i.e., applying the Svedberg relationship. It was demonstrated that the use of linear PEG for the SEC calibration results in the significantly underestimated values of the molar masses of hbPEGs. The largest hbPEG samples exhibited a hydrodynamic radius of ≈14 nm in aqueous solution. The obtained Kuhn–Mark–Houwink–Sakurada scaling relations show linear trends in all range of molar masses. The detected scaling indexes virtually correspond to the homologous series characterized by a direct proportionality between the molar mass and the volume of the macromolecules that make up this series. The effect of branching on the molecular dimensions and on the hydrodynamic characteristics is discussed, and the corresponding contraction factors have been estimated.
Co-reporter:Eva-Maria Christ, Dominika Hobernik, Matthias Bros, Manfred Wagner, and Holger Frey
Biomacromolecules 2015 Volume 16(Issue 10) pp:
Publication Date(Web):September 10, 2015
DOI:10.1021/acs.biomac.5b00951
The cationic ring-opening copolymerization of 3,3-bis(hydroxymethyl)oxetane (BHMO) with glycidol using different comonomer ratios (BHMO content from 25 to 90%) and BF3OEt2 as an initiator has been studied. Apparent molecular weights of the resulting hyperbranched polyether copolymers ranged from 1400 to 3300 g mol–1 (PDI: 1.21–1.48; method: SEC, linear PEG standards). Incorporation of both comonomers is evidenced by MALDI-TOF mass spectroscopy. All hyperbranched polyether polyols with high content of primary hydroxyl groups portray good solubility in water, which correlates with an increasing content of glycerol units. Detailed NMR characterization was employed to elucidate the copolymer microstructures. Kinetic studies via FTIR demonstrated a weak gradient-type character of the copolymers. MTT assays of the copolymers (up to 100 μg mL–1) on HEK and fibroblast cell lines (3T3, L929, WEHI) as well as viability tests on the fibroblast cells were carried out to assess the biocompatibility of the materials, confirming excellent biocompatibility. Transfection efficiency characterization by flow cytometry and confocal laser microscopy demonstrated cellular uptake of the copolymers. Antiadhesive properties of the materials on surfaces were assessed by adhesion assays with fibroblast cells.
Co-reporter:Hannah Pohlit, Iris Bellinghausen, Martina Schömer, Bärbel Heydenreich, Joachim Saloga, and Holger Frey
Biomacromolecules 2015 Volume 16(Issue 10) pp:
Publication Date(Web):August 31, 2015
DOI:10.1021/acs.biomac.5b00458
In the last decades, the number of allergic patients has increased dramatically. Allergen-specific immunotherapy (SIT) is the only available cause-oriented therapy so far. SIT reduces the allergic symptoms, but also exhibits some disadvantages; that is, it is a long-lasting procedure and severe side effects like anaphylactic shock can occur. In this work, we introduce a method to encapsulate allergens into nanoparticles to avoid severe side effects during SIT. Degradable nanocarriers combine the advantage of providing a physical barrier between the encapsulated cargo and the biological environment as well as responding to certain local stimuli (like pH) to release their cargo. This work introduces a facile strategy for the synthesis of acid-labile poly(ethylene glycol) (PEG)-macromonomers that degrade at pH 5 (physiological pH inside the endolysosome) and can be used for nanocarrier synthesis. The difunctional, water-soluble PEG dimethacrylate (PEG-acetal-DMA) macromonomers with cleavable acetal units were analyzed with 1H NMR, SEC, and MALDI-ToF-MS. Both the allergen and the macromonomers were entrapped inside liposomes as templates, which were produced by dual centrifugation (DAC). Radical polymerization of the methacrylate units inside the liposomes generated allergen-loaded PEG nanocarriers. In vitro studies demonstrated that dendritic cells (DCs) internalize the protein-loaded, nontoxic PEG-nanocarriers. Furthermore, we demonstrate by cellular antigen stimulation tests that the nanocarriers effectively shield the allergen cargo from detection by immunoglobulins on the surface of basophilic leucocytes. Uptake of nanocarriers into DCs does not lead to cell maturation; however, the internalized allergen was capable to induce T cell immune responses.
Co-reporter:Jana Herzberger and Holger Frey
Macromolecules 2015 Volume 48(Issue 22) pp:8144-8153
Publication Date(Web):November 5, 2015
DOI:10.1021/acs.macromol.5b02178
Both homo- and copolymerization of the hitherto nonpolymerizable epoxide monomer epicyanohydrin (EPICH) with ethylene oxide (EO) have been studied, employing the monomer activation technique. Tetraoctylammonium bromide or tetrabutylammonium iodide was used as initiator combined with i-Bu3Al to activate the EPICH monomer. The EPICH content was varied from 4 to 16 mol %, yielding well-defined PEG-co-PEPICH copolymers with molecular weights Mn (SEC) ranging from 3700 to 8800 g mol–1. The nitrile groups of the resulting polyethers were further reduced or hydrolyzed to introduce amino, amide, or carboxyl groups at the polyether backbone, circumventing protecting group chemistry. Successful transformation of the functional groups was proven by SEC measurements, 1H NMR, 13C NMR, and FT-IR spectroscopy. These carboxyl-functional PEG copolymers are anionic polyelectrolytes consisting only of purely aliphatic polyether structures with carboxyl groups. The hydrolyzed PEPICH homopolymers represent the first polyether-based analogues of poly(acrylamide) and poly(acrylic acid). In pronounced contrast to poly(acrylic acid), carboxyl-functional PEPICH shows excellent aqueous solubility even at low pH.
Co-reporter:Achim T. Reibel, Sophie S. Müller, Stefanie Pektor, Nicole Bausbacher, Matthias Miederer, Holger Frey, and Frank Rösch
Biomacromolecules 2015 Volume 16(Issue 3) pp:
Publication Date(Web):February 3, 2015
DOI:10.1021/bm5017332
In this study, linear poly(ethylene glycol) (PEG) and novel linear-hyperbranched, amphiphilic polyglycerol (hbPG) polymers with cholesterol (Ch) as a lipid anchor moiety were radiolabeled with fluorine-18 via copper-catalyzed click chemistry. In vivo investigations via positron emission tomography (PET) and ex vivo biodistribution in mice were conducted. A systematic comparison to the liposomal formulations with and without the polymers with respect to their initial pharmacokinetic properties during the first hour was carried out, revealing remarkable differences. Additionally, cholesterol was directly labeled with fluorine-18 and examined likewise. Both polymers, Ch-PEG27-CH2-triazole-TEG-18F and Ch-PEG30-hbPG24-CH2-triazole-TEG-18F (TEG: triethylene glycol), showed rapid renal excretion, whereas the 18F-cholesten displayed retention in lung, liver, and spleen. Liposomes containing Ch-PEG27-CH2-triazole-TEG-18F revealed a hydrodynamic radius of 46 nm, liposomal Ch-PEG30-hbPG24-CH2-triazole-TEG-18F showed a radius of 84 nm and conventional liposomes with 18F-cholesten 204 nm, respectively. The results revealed fast uptake of the conventional liposomes by liver, spleen, and lung. Most importantly, the novel hbPG-polymer stabilized liposomes showed similar behavior to the PEG-shielded vesicles. Thus, an advantage of multifunctionality is achieved with retained pharmacokinetic properties. The approach expands the scope of polymer tracking in vivo and liposome tracing in mice via PET.
Co-reporter:Jeannette Hilf, Andrew Phillips and Holger Frey
Polymer Chemistry 2014 vol. 5(Issue 3) pp:814-818
Publication Date(Web):11 Sep 2013
DOI:10.1039/C3PY00977G
Functional poly(carbonate)s with multiple hydroxyl functionalities have been prepared by copolymerization of carbon dioxide (CO2) with glycidyl methyl ether (GME) and benzyl glycidyl ether (BGE) in various ratios, using a diethylzinc–pyrogallol catalyst system. Subsequent catalytic hydrogenation was employed for removal of the benzyl protecting groups at the polymer backbone. A series of copolymers with varying comonomer fractions from 0 to 100% was obtained. The copolymers possessed a broad range of molecular weights from 9000 to 30000 g mol−1 and showed polydispersities Mw/Mn between 2.4 and 3.6. The materials were characterized via1H and 13C NMR, SEC and differential scanning calorimetry (DSC). The deprotected copolymers are structurally simple materials consisting of carbon dioxide, GME and glycerol. These functional poly(carbonate)s represent degradable materials with tailored functionality.
Co-reporter:Adrian Natalello, Arda Alkan, Philipp von Tiedemann, Frederik R. Wurm, and Holger Frey
ACS Macro Letters 2014 Volume 3(Issue 6) pp:560
Publication Date(Web):May 30, 2014
DOI:10.1021/mz500255h
The functional group distribution along the polymer backbone resulting from the living anionic copolymerization of styrene (S) and para-but-3-enyl styrene (pBuS) was investigated in cyclohexane at room temperature. A variety of copolymers with different comonomer contents x(S) = 0–0.84 have been synthesized with molecular weight dispersities Mw/Mn ≤1.12. All polymers have been characterized in detail by 1H NMR spectroscopy, size exclusion chromatography (SEC), and differential scanning calorimetry (DSC). A detailed understanding of the monomer sequence distribution during the copolymerization was achieved by real-time 1H NMR spectroscopy. This technique permits us to determine the changing monomer concentration of each monomer in stock throughout the reaction. Consequently, monomer incorporation and thus the probability of incorporation can be determined at any time of the copolymerization, and a precise determination of the functional group density along the polymer chain is possible. To demonstrate accessibility of the olefin side chains of the copolymer for transformations, quantitative thiol–ene addition of a cysteine derivative has been studied.
Co-reporter:Christian Moers;Robert Wrazidlo;Adrian Natalello;Isabelle Netz;Mihail Mondeshki
Macromolecular Rapid Communications 2014 Volume 35( Issue 11) pp:1075-1080
Publication Date(Web):
DOI:10.1002/marc.201400017
Co-reporter:Jeannette Hilf;Patricia Schulze;Jan Seiwert
Macromolecular Rapid Communications 2014 Volume 35( Issue 2) pp:198-203
Publication Date(Web):
DOI:10.1002/marc.201300663
Co-reporter:Adrian Natalello, Jan Morsbach, Andreas Friedel, Arda Alkan, Christoph Tonhauser, Axel H. E. Müller, and Holger Frey
Organic Process Research & Development 2014 Volume 18(Issue 11) pp:1408-1412
Publication Date(Web):July 21, 2014
DOI:10.1021/op500149t
We describe the living anionic polymerization of 2-vinylpyridine (2VP) and styrene (S) in continuous flow, comparing two micromixing devices with different mixing principles. The use of a continuous flow setup reduces the experimental effort for living anionic polymerizations significantly, compared to a conventional batch system. By adjusting the ratio of the flow rates of the monomer and initiator solutions a variety of different molecular weights can be rapidly synthesized within several minutes, using one setup. Additionally, a comparison of the influence of the two different mixing devices—an interdigital micromixer (SIMM-V2) leading to laminar mixing and a tangential four-way jet mixing device leading to a turbulent mixing pattern—has been achieved. Both setups allow living anionic polymerization in polar solvents at room temperature with full monomer conversion within seconds and yield polymers with narrowly distributed molecular weights. A maximum Mn of approximately 149,000 g mol–1 (PS-9, PDI = 1.04) for PS and 96,000 g mol–1 for P2VP (P2VP-15, PDI = 1.05) was obtained. Clearly, the turbulent four-way jet mixing device led to lower polydispersity than the laminar mixing device. All polymers were characterized by 1H NMR spectroscopy and size exclusion chromatography (SEC).
Co-reporter:Muhammet U. Kahveci;Christine Mangold;Yusuf Yagci
Macromolecular Chemistry and Physics 2014 Volume 215( Issue 6) pp:566-571
Publication Date(Web):
DOI:10.1002/macp.201300794
Co-reporter:Sophie S. Müller, Christian Moers, and Holger Frey
Macromolecules 2014 Volume 47(Issue 16) pp:5492-5500
Publication Date(Web):August 12, 2014
DOI:10.1021/ma501280k
Motivated by the oxygen-rich and fully amorphous structure of poly(glycidyl methyl ether) (PGME), a series of thermoresponsive poly(glycidyl methyl ether-co-ethylene oxide) copolymers P(GME-co-EO) with molecular weights in the range of 3000–20 000 g mol–1 were synthesized by the activated monomer polymerization technique. Tetraoctylammonium bromide (NOct4Br) was employed as an initiator in combination with triisobutylaluminum (i-Bu3Al) as a catalyst under mild conditions. Polyethers with varying GME content between 31 and 100 mol % were obtained. Triad sequence analysis using 13C NMR spectroscopy proved that no pronounced block structure was obtained. Differential scanning calorimetry (DSC) revealed that samples exceeding 65 mol % content of GME are amorphous, whereas with lower GME content a low degree of crystallization was observed. Melting temperatures for these polyethers were in the range 9.8–37.5 °C. Furthermore, the copolymers’ lower critical solution temperatures (LCSTs) in aqueous solution were tuned from 55 °C for the PGME homopolymer up to 98 °C by varying the amount of GME. The approach permits to combine two highly biocompatible and water-soluble materials.
Co-reporter:Jana Herzberger, Dennis Kurzbach, Mathias Werre, Karl Fischer, Dariush Hinderberger, and Holger Frey
Macromolecules 2014 Volume 47(Issue 22) pp:7679-7690
Publication Date(Web):November 3, 2014
DOI:10.1021/ma501367b
Amine-functional poly(ethylene glycol) (PEG) copolymers have been prepared that exhibit thermo- and pH- responsive behavior in aqueous solution. Three novel tertiary di(n-alkyl)glycidylamine monomers have been introduced for anionic ring-opening copolymerization (AROcP) with ethylene oxide (EO): N,N-di(n-butyl)glycidylamine (DButGA), N,N-di(n-hexyl)glycidylamine (DHexGA), and N,N-di(n-octyl)glycidylamine (DOctGA). Via controlled AROcP we synthesized well-defined (Mw/Mn = 1.05–1.14), water-soluble block- and gradient-type PEG copolymers, containing up to 25 mol % of the respective dialkylglycidylamine comonomer. Molecular weights ranged from 4900 to 12 000 g mol–1. Detailed in-situ 1H NMR kinetics and 13C triad analyses elucidate the microstructures of the copolymers and the relative reactivity of the novel comonomers. Notably, the n-alkyl chain length had no significant influence on the relative reactivity of the glycidylamine comonomers. Calculated reactivity ratios ranged from rEO = 1.84, rDButGA = 0.49 to rEO = 1.78, rDOctGA = 0.42, manifesting the formation of gradient copolymers. Thermo- and pH-responsive properties of these copolymers are precisely tunable by the comonomer ratio, and cloud points in aqueous solution can be adjusted between 21 and 93 °C. Electron paramagnetic resonance (EPR) spectroscopic studies with TEMPO as a spin probe were conducted to elucidate host–guest interactions of the copolymers. Unexpectedly, the n-alkyl chain length of the different glycidylamine comonomers only influences the inverse phase transition of the gradient copolymers, but not of the block copolymers on the nanoscale. Self-assembly of the block- and gradient-type copolymers in aqueous alkaline solution by both static and dynamic light scattering has also been investigated after confirming the existence of pure unimers in methanol.
Co-reporter:Eva-Maria Christ;Sophie S. Müller;Elena Berger-Nicoletti
Journal of Polymer Science Part A: Polymer Chemistry 2014 Volume 52( Issue 19) pp:2850-2859
Publication Date(Web):
DOI:10.1002/pola.27315
ABSTRACT
Synthesis and characterization of novel hydroxyl-functionalized oxetane-inimers with varied alkyl chain length—3-hydroxymethyl-3-methoxymethyloxetane, 3-hydroxymethyl-3-propoxymethyloxetane, and 3-hexoxymethyl-3-hydroxymethyloxetane—is reported. Cationic ring-opening polymerization of these latent, cyclic AB2-monomers leads to hyperbranched (hb) polyether polyols with degrees of branching between 34 and 69%, confirmed by inverse-gated (IG) 13C NMR spectroscopy. The hyperbranching polymerization yielded apparent molecular weights (Mn) ranging from 500 to 2500 g mol−1 (size exclusion chromatography). Remarkably, by copolymerization of 1,1,1-tris(4-hydroxyphenyl)ethane as a “focal” unit, polymerization under slow monomer addition conditions lead to higher apparent molecular weights up to 11,220 g mol−1. The end groups of the hb polymers were studied via matrix-assisted laser desorption/ionization time of flight mass and NMR spectrometry. By varying the alkyl chain length, tailoring of the solubility and glass transition temperatures of the materials is possible. Potential applications range from macroinitiators with defined polarity to tailoring of surface properties of antifouling materials. © 2014 Wiley Periodicals, Inc. J. Polym. Sci., Part A: Polym. Chem. 2014, 52, 2850–2859
Co-reporter:Markus Glaffig;Björn Palitzsch;Sebastian Hartmann;Dr. Christoph Schüll;Lutz Nuhn;Dr. Bastian Gerlitzki;Dr. Edgar Schmitt;Dr. Holger Frey;Dr. Horst Kunz
Chemistry - A European Journal 2014 Volume 20( Issue 15) pp:4232-4236
Publication Date(Web):
DOI:10.1002/chem.201400256
Abstract
For antitumor vaccines both the selected tumor-associated antigen, as well as the mode of its presentation, affect the immune response. According to the principle of multiple antigen presentation, a tumor-associated MUC1 glycopeptide combined with the immunostimulating T-cell epitope P2 from tetanus toxoid was coupled to a multi-functionalized hyperbranched polyglycerol by “click chemistry”. This globular polymeric carrier has a flexible dendrimer-like structure, which allows optimal antigen presentation to the immune system. The resulting fully synthetic vaccine induced strong immune responses in mice and IgG antibodies recognizing human breast-cancer cells.
Co-reporter:Adrian Natalello, Christoph Tonhauser, and Holger Frey
ACS Macro Letters 2013 Volume 2(Issue 5) pp:409
Publication Date(Web):April 26, 2013
DOI:10.1021/mz400147z
Living anionic polymerization of para-(1-ethoxy ethoxy)styrene (pEES) resulting in molecular weights between 2700 and 69 000 g mol–1 and polydispersity indices ≤1.09 is introduced. PpEES can be used as a precursor for the synthesis of well-defined poly(p-hydroxystyrene) (PHS) architectures, enabling facile and rapid acidic deprotection at room temperature within a few minutes. In addition, a series of block copolymers containing pEES and 2-vinylpyridine (2VP) have been synthesized by anionic block copolymerization, with varied block ratios (X2VP) between 0.13 and 0.83. Characterization by 1H NMR spectroscopy, size exclusion chromatography (SEC), and differential scanning calorimetry (DSC) was carried out, and all polymers have been deprotected, leading to the respective PHS-b-P2VP block copolymers. Furthermore, PHS-b-P2VP has been used as a macroinitiator for the anionic ring-opening polymerization of ethylene oxide (EO) to generate ((PHS-g-PEO51)13-b-P2VP40) graft-block copolymers.
Co-reporter:Christoph Schüll, Tile Gieshoff and Holger Frey
Polymer Chemistry 2013 vol. 4(Issue 17) pp:4730-4736
Publication Date(Web):08 Jul 2013
DOI:10.1039/C3PY00707C
By copolymerization of glycidol with the alkyne-containing oxirane monomer glycidyl propargyl ether (GPE), hyperbranched polyglycerol (hbPG) with a defined number of alkyne functionalities (up to 38%) can be obtained in a one-step procedure. The number of alkynes can be adjusted by the glycidol/GPE ratio to provide multi-alkyne functional hbPGs, maintaining the highly branched polyether structure. Interestingly, the acidic proton of the alkyne moiety does not interfere with the proton exchange mechanism during the polymerization of glycidol. By specific modification of the synthesis procedure, crosslinking reactions can be suppressed. The polymers exhibit molecular weights ranging from 1800 to 5500 g mol−1 (determined by 1H NMR spectroscopy and SEC) with moderate polydispersities (Mw/Mn < 2.0, mostly <1.7). Using inverse gated 13C NMR spectroscopy and two-dimensional NMR techniques, the different repeat units of the copolymers can be assigned. The degree of branching value (DB) ranges from 0.58 to 0.50 for increasing GPE content, which is caused by an increased number of linear repeat units with increasing GPE content. The alkyne functionalities are readily available for derivatization reactions by copper-catalyzed azide–alkyne “click”-type cycloaddition reactions. The convenient synthesis and the broad applicability of the alkyne functionalities render these copolymers interesting building blocks for the preparation of complex polymer architectures by “click”-chemistry. This is exemplified by the attachment of hydrophobic azide end-functional polystyrene to yield amphiphilic branched copolymers containing exactly one hbPG block.
Co-reporter:Jeannette Hilf
Macromolecular Rapid Communications 2013 Volume 34( Issue 17) pp:1395-1400
Publication Date(Web):
DOI:10.1002/marc.201300425
Co-reporter:Jeannette Geschwind
Macromolecular Rapid Communications 2013 Volume 34( Issue 2) pp:150-155
Publication Date(Web):
DOI:10.1002/marc.201200682
Abstract
A series of functional polycarbonates, poly((isopropylidene glyceryl glycidyl ether)-co-(glycidyl methyl ether) carbonate) (P((IGG-co-GME) C)) random copolymers with different fractions of 1,2-isopropylidene glyceryl glycidyl ether (IGG) units, is synthesized. After acidic hydrolysis of the acetal protecting groups, a new type of functional polycarbonate prepared directly from CO2 and glycerol is obtained, namely poly((glyceryl glycerol)-co-(glycidyl methyl ether) carbonate) (P((GG-co-GME) C)). All hydroxyl functional samples exhibit monomodal molecular weight distributions with PDIs between 2.5 and 3.3 and Mn between 12 000 and 25 000 g mol−1. Thermal properties reflect the amorphous structure of the polymers. The materials are stable in bulk and solution.
Co-reporter:Jeannette Geschwind;Sahas Rathi;Christine Tonhauser;Martina Schömer;Shaw Ling Hsu;E. Bryan Coughlin
Macromolecular Chemistry and Physics 2013 Volume 214( Issue 13) pp:1434-1444
Publication Date(Web):
DOI:10.1002/macp.201300074
Co-reporter:Jeannette Hilf;Patricia Schulze
Macromolecular Chemistry and Physics 2013 Volume 214( Issue 24) pp:2848-2855
Publication Date(Web):
DOI:10.1002/macp.201300586
Co-reporter:Jeannette Geschwind;Frederik Wurm
Macromolecular Chemistry and Physics 2013 Volume 214( Issue 8) pp:892-901
Publication Date(Web):
DOI:10.1002/macp.201200608
Co-reporter:Adrian Natalello, Mathias Werre, Arda Alkan, and Holger Frey
Macromolecules 2013 Volume 46(Issue 21) pp:8467-8471
Publication Date(Web):November 1, 2013
DOI:10.1021/ma401847y
Detailed understanding of the monomer sequence distribution in carbanionic copolymerization was achieved by direct online monitoring of copolymerizations in an NMR tube. Obtaining detailed knowledge of the changing monomer concentration in stock during the reaction, this technique permits to determine the incorporation probability for each monomer at every position of the polymer chain. An in situ kinetic study of two different carbanionic copolymerizations has been carried out. On the one hand, the copolymerization of the structurally similar, protected hydroxystyrene derivatives, p-(1-ethoxy ethoxy)styrene (pEES) and 4-tert-butoxystyrene (tBuOS), and on the other hand the copolymerization of the chemically different monomers, styrene (S) and pEES, have been studied. Whereas in the first case a slight deviation from an ideal random copolymerization was observed, the latter copolymerization leads to gradient copolymers. Real-time 1H NMR spectroscopy gave detailed insight into the reaction behavior at every stage of the copolymerization and leads to precise understanding of the resulting gradient structures.
Co-reporter:Anna M. Fischer, Raphael Thiermann, Michael Maskos, Holger Frey
Polymer 2013 Volume 54(Issue 8) pp:1993-2000
Publication Date(Web):3 April 2013
DOI:10.1016/j.polymer.2012.12.044
Using a hyperbranched poly(glycolide) (hbPGA) macroinitiator the synthesis of poly(l-lactide) (PLLA) multi-arm star polyesters has been achieved via a core-first approach. The star-shaped copolymers were prepared in a one-pot two-step process via Sn(Oct)2-catalyzed ring-opening polymerization (ROP) conducted in the melt. Complete conversion of the end groups of the hbpolyglycolide polyester polyols is ensured by the reactive primary hydroxyl termini. By adjusting the monomer/initiator ratio a series of star copolymers with varying PLLA arm length has been obtained with molecular weights in the range of 1500 to 10,000 g/mol (SEC). The successful coupling of the PLLA arms to the hbPGA core has been confirmed via detailed 1D and 2D NMR spectroscopy. Because of the different hydrodynamic volume of the star polymers in contrast to their linear analogs, the weight-average molecular weight (Mw) was determined both by SEC and static light scattering (SLS). The star-shaped poly(lactide)s reveal different thermal properties in comparison to linear poly(lactide) homopolymers.
Co-reporter:Christoph Schüll, Hauke Rabbel, Friederike Schmid, and Holger Frey
Macromolecules 2013 Volume 46(Issue 15) pp:5823-5830
Publication Date(Web):July 29, 2013
DOI:10.1021/ma401119r
The hypergrafting strategy designates the synthesis of hyperbranched graft copolymers (HGCs) in a grafting-from approach, using ABm monomers, from multifunctional, polydisperse macroinitiator cores by slow monomer addition. Hypergrafting leads to complex polymer topologies with defined molecular weight, degree of branching (DB), and polydispersity (PD). By a generating function formalism, a generally applicable equation for the PD of HGCs (PD = PDf + (m – 1)/f̅) is derived, where PDf is the polydispersity of the core and f̅ its average functionality. In addition, the complete molecular weight distribution function has been calculated for varied m and f̅ as well as for a given distribution of initiator functionalities f. For comparison of the theoretical predictions with experimental results, a series of novel linear polyglycerol-graft-hyperbranched polyglycerol (linPG-g-hbPG) HGCs (Mn = 1000–4000 g mol–1) were synthesized and characterized as a model system. An increase in polydispersity occurred as a consequence of the hypergrafting process, confirming the theoretical predictions of the novel equation. Moreover, the model system allows for the determination of the DB of hbPG prepared by hypergrafting from linear polyglycerol macroinitatiors (DB = 0.59–0.61). The theoretical results presented are key to achieve control over the branch-on-branch topology of hyperbranched blocks in nonconventional polymer architectures, such as linear–hyperbranched block copolymers.
Co-reporter:Christian Moers, Lutz Nuhn, Marcel Wissel, René Stangenberg, Mihail Mondeshki, Elena Berger-Nicoletti, Anja Thomas, David Schaeffel, Kaloian Koynov, Markus Klapper, Rudolf Zentel, and Holger Frey
Macromolecules 2013 Volume 46(Issue 24) pp:9544-9553
Publication Date(Web):December 3, 2013
DOI:10.1021/ma402081h
Complex, reversible hyperbranched graft polymer topologies have been obtained by spontaneous self-assembly. Well-defined adamantyl- and β-cyclodextrin-functionalized polymers were employed to generate linear-g-(linear–hyperbranched) supramolecular graft terpolymers. For this purpose the synthesis of monoadamantyl-functionalized linear polyglycerols (Ada-linPG) and hyperbranched polyglycerols (Ada-hbPG) as well as poly(ethylene glycol)-block-linear polyglycerol (Ada-PEG-b-linPG) and poly(ethylene glycol)-block-hyperbranched poly(glycerol) (Ada-PEG-b-hbPG) block copolymers was established. Isothermal titration calorimetry (ITC) with β-cyclodextrin revealed a shielding effect of hyperbranched polyglycerol for the adamantyl functionality, which was significantly less pronounced when using a linear spacer chain between the adamantyl residue and the hyperbranched polyglycerol block. Additionally, well-defined poly(2-hydroxypropylamide) (PHPMA) with pendant β-cyclodextrin moieties was synthesized via RAFT polymerization and sequential postpolymerization modification. Upon mixing of the β-cyclodextrin-functionalized PHPMA with Ada-PEG-b-hbPG, a supramolecular linear-g-(linear–hyperbranched) graft terpolymer was formed. The self-assembly was proven by ITC, diffusion-ordered NMR spectroscopy (DOSY), and fluorescence correlation spectroscopy (FCS).
Co-reporter:Jeannette Geschwind and Holger Frey
Macromolecules 2013 Volume 46(Issue 9) pp:3280-3287
Publication Date(Web):April 29, 2013
DOI:10.1021/ma400090m
The functional, aliphatic poly(1,2-glycerol carbonate) as a fundamental, simple polymer structure based on glycerol and CO2 was prepared by combination of glycidyl ether monomers with carbon dioxide via two different approaches. The material was obtained by two-step procedures either via copolymerization of (i) ethoxy ethyl glycidyl ether (EEGE) or (ii) benzyl glycidyl ether (BGE) with CO2, followed by removal of the respective protecting groups via acidic cleavage for (i) and hydrogenation for (ii). The resulting protected polycarbonate structures and the targeted poly(1,2-glycerol carbonate) were investigated with 1H NMR and 13C NMR spectroscopy as well as 2D-NMR methods. Removal of both protecting groups was possible without significant backbone degradation; however, the hydrogenation route for (ii) turned out to be advantageous. All new poly(carbonate)s have been characterized with respect to their thermal behavior. Protected and deprotected poly(1,2-glycerol carbonate)s were obtained with molecular weights in the range of 5000–25 200 g/mol and a PDI from 1.24 to 2.33. The degradation kinetics of poly(1,2-glycerol carbonate) in DMF has also been studied, demonstrating rather rapid degradation within several days to oligomers and cyclic carbonates.
Co-reporter:Rebecca Klein, Christoph Schüll, Elena Berger-Nicoletti, Michael Haubs, Klaus Kurz, and Holger Frey
Macromolecules 2013 Volume 46(Issue 22) pp:8845-8852
Publication Date(Web):November 14, 2013
DOI:10.1021/ma4015565
The synthesis of hyperbranched-linear-hyperbranched ABA triblock copolymers based on a linear poly(oxymethylene) (POM) block and hyperbranched poly(glycerol) (hbPG) blocks is described. The polymers containing a polyacetal polyether structure were prepared from linear bishydroxy-functional POM macroinitiators, obtained by cationic ring-opening polymerization of trioxane and 1,3-dioxolane as a comonomer with formic acid as a transfer agent and subsequent hydrolysis of the formate group. Partial deprotonation of the resulting hydroxyl groups permitted “hypergrafting” of glycidol by anionic ring-opening multibranching polymerization (ROMBP). With respect to the hyperbranched blocks, the obtained polymers show the expected, moderate molecular weight distributions in the range of 1.31 to 2.01 and molecular weights between 6100 and 22900 g mol–1. Both the degree of polymerization of the linear POM segments as well as the molecular weight of the hbPG blocks have been varied. Key properties, such as thermal behavior and thermal stability, degree of crystallization and surface properties were investigated, showing an adjustable degree of crystallization and enhanced hydrophilicity depending on the hbPG content.
Co-reporter:Christoph Schüll, Holger Frey
Polymer 2013 Volume 54(Issue 21) pp:5443-5455
Publication Date(Web):4 October 2013
DOI:10.1016/j.polymer.2013.07.065
In this feature article, the grafting of hyperbranched polymers to different substrates is reviewed. Both grafting onto macromolecules with different topologies (homogeneous grafting) and the resulting complex polymer architectures containing highly branched segments as well as their applications are discussed. In the second part grafting of hyperbranched polymers on surfaces, i.e., planar surfaces and spherical particles (heterogeneous grafting), with respect to specific applications, such as bio-repellent surfaces or soluble carbon nanotubes is described. In all cases, the one-step synthesis and the resulting highly branched topology of the hyperbranched building blocks is beneficial for the convenient introduction of a large number of functional groups to the substrates. These multifunctional hybrid materials open interesting options for applications, e.g., for highly functional nanoparticles or nanocomposites.Figure optionsDownload full-size imageDownload as PowerPoint slide
Co-reporter:Martina Schömer;Christoph Schüll
Journal of Polymer Science Part A: Polymer Chemistry 2013 Volume 51( Issue 5) pp:995-1019
Publication Date(Web):
DOI:10.1002/pola.26496
Abstract
Hyperbranched polymers, dendritic macromolecules with branch-on-branch structures, have become an important polymer class since the early 1990s. They combine several advantages of the perfectly branched dendrimers with easy accessibility, typically in a one-step synthesis. Hyperbranched polyethers are a particularly interesting class of chemically stable and often biocompatible materials. Multifunctional hyperbranched polyethers with controllable molar mass and comparably low polydispersities can been prepared using hydroxyl-functional epoxides or oxetanes for polymerization via anionic and cationic polymerization mechanisms. Here, we review the progress in the preparation, characterization, and application of these uniquely versatile aliphatic polyether polyols. Their unusual mechanical, thermal, and solution properties render them useful for a variety of applications, for example, as building blocks for various complex macromolecular architectures or in biomedical applications. © 2012 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem, 2013
Co-reporter:Valerie S. Wilms, Heiko Bauer, Christine Tonhauser, Anna-Maria Schilmann, Marc-Christian Müller, Wolfgang Tremel, and Holger Frey
Biomacromolecules 2013 Volume 14(Issue 1) pp:
Publication Date(Web):December 4, 2012
DOI:10.1021/bm3015889
Bifunctional CA-PEG (catechol-poly(ethylene glycol)) and multifunctional CA-PEG-PGA/PEVGE (poly(glycidyl amine)/poly(ethylene glycol vinyl glycidyl ether)) ligands for the functionalization and solubilization of nanoparticles are introduced. Tunable polymers with polydispersities <1.25 and molecular weights in the range 500–7700 g mol–1 containing a catechol moiety for conjugation to metal oxide nanoparticles were prepared. The functional PEG ligands were synthesized starting from the acetonide-protected catechol initiator 2,2-dimethyl-1,3-benzodioxole-5-propanol (CA-OH) for oxyanionic polymerization. CA-OH was used both for homopolymerization of ethylene oxide (EO) as well as copolymerization with functional epoxides N,N-diallyl glycidyl amine (DAGA), releasing primary amino groups and ethylene glycol vinyl glycidyl ether (EVGE), exhibiting a double bond for click-type reactions, to generate CA-PEG and CA-PEG-PGA/PEVGE. We demonstrate the potential of the functional ligands by binding to MnO nanoparticles, rendering the PEGylated nanoparticles highly stable in aqueous environment. Furthermore, addressability of the functional groups has been proven, for example, by coupling with fluoresceine isothiocyanate (FITC), to allow for optical monitoring of the nanoparticle fate in biological systems.
Co-reporter:Carsten Dingels, Sophie S. Müller, Tobias Steinbach, Christine Tonhauser, and Holger Frey
Biomacromolecules 2013 Volume 14(Issue 2) pp:
Publication Date(Web):December 20, 2012
DOI:10.1021/bm3016797
Poly(ethylene glycol) (PEG) with acid-sensitive moieties gained attention particularly for various biomedical applications, such as the covalent attachment of PEG (PEGylation) to protein therapeutics, the synthesis of stealth liposomes, and polymeric carriers for low-molecular-weight drugs. Cleavable PEGs are favored over their inert analogues because of superior pharmacodynamic and/or pharmacokinetic properties of their formulations. However, synthetic routes to acetal-containing PEGs published up to date either require enormous efforts or result in ill-defined materials with a lack of control over the molecular weight. Herein, we describe a novel methodology to implement a single acetaldehyde acetal in well-defined (hetero)functional poly(ethylene glycol)s with total control over its position. To underline its general applicability, a diverse set of initiators for the anionic polymerization of ethylene oxide (cholesterol, dibenzylamino ethanol, and poly(ethylene glycol) monomethyl ether (mPEG)) was modified and used to synthesize the analogous labile PEGs. The polyether bearing the cleavable lipid had a degree of polymerization of 46, was amphiphilic and exhibited a critical micelle concentration of 4.20 mg·L–1. From dibenzylamino ethanol, three heterofunctional PEGs with different molecular weights and labile amino termini were generated. The transformation of the amino functionality into the corresponding squaric acid ester amide demonstrated the accessibility of the cleavable functional group and activated the PEG for protein PEGylation, which was exemplarily shown by the attachment to bovine serum albumin (BSA). Furthermore, turning mPEG into a macroinitiator with a cleavable hydroxyl group granted access to a well-defined poly(ethylene glycol) derivative bearing a single cleavable moiety within its backbone. All the acetal-containing PEGs and PEG/protein conjugates were proven to degrade upon acidic treatment.
Co-reporter:Christoph Schüll and Holger Frey
ACS Macro Letters 2012 Volume 1(Issue 4) pp:461
Publication Date(Web):March 20, 2012
DOI:10.1021/mz200250s
Linear polymers with hyperbranched side chains are unusual macromolecular structures due to their high number of functional groups in the side chains as well as their potential cylindrical conformation in bulk or solution. In a three-step synthesis combining anionic and oxy-anionic polymerization, hyperbranched polyglycerol was “hypergrafted” from linear poly(4-hydroxy styrene) macroinitiators to yield poly(4-hydroxy styrene)-graft-hyperbranched polyglycerol. Successful grafting with control over molecular weight (10–31 kg·mol–1) and low PDIs (<1.4) was shown by various characterization techniques. All polymers have a high side chain density, due to rapid transfer of the initiating functional groups to the linear backbone. DSC studies give insight into the thermal properties of the resulting polymers.
Co-reporter:Martina Schömer, Jan Seiwert, and Holger Frey
ACS Macro Letters 2012 Volume 1(Issue 7) pp:888
Publication Date(Web):June 29, 2012
DOI:10.1021/mz300256y
Backbone-thermoresponsive hyperbranched poly(propylene oxide)-based polyether polyols have been synthesized by anionic ring-opening copolymerization of glycidol and propylene oxide. The number of functional hydroxyl end groups and the lower critical solution temperature (LCST) can be readily adjusted by varying the comonomer ratio. Molecular weights in the range of 1200–2000 g/mol were achieved. Hyperbranched polyether polyols with LCST values between 24 and 83 °C can be obtained in a convenient one-step reaction.
Co-reporter:Christine Mangold, Frederik Wurm and Holger Frey
Polymer Chemistry 2012 vol. 3(Issue 7) pp:1714-1721
Publication Date(Web):12 Jan 2012
DOI:10.1039/C2PY00489E
In this review article functional epoxide monomers that are suitable for controlled ring-opening polymerization (ROP) are discussed. Functional epoxides possess reactive groups, which either are directly accessible or carry suitable protective groups that can be removed in a facile one-step reaction after polymerization. The methods used to obtain linear, functional aliphatic polyethers rely on living polymerization techniques for the synthesis of well-defined structures. Materials properties, such as thermo-responsive behavior in combination with different functional groups that can be addressed selectively, render these novel materials interesting for a variety of applications.
Co-reporter:Christine Tonhauser, Christoph Schüll, Carsten Dingels, and Holger Frey
ACS Macro Letters 2012 Volume 1(Issue 9) pp:1094
Publication Date(Web):August 15, 2012
DOI:10.1021/mz300265z
The introduction of acid-degradable acetal moieties into a hyperbranched polyether backbone has been achieved by the design of a novel epoxide-based degradable inimer. This new monomer, namely, 1-(glycidyloxy)ethyl ethylene glycol ether (GEGE), has been copolymerized in the anionic ring-opening polymerization (AROP) with ethylene oxide (EO) or glycidol (G), respectively, yielding branched polyethers, that is, P(EO-co-GEGE) and P(G-co-GEGE), that possess an adjustable amount of acid-cleavable acetal units. In addition, a novel class of multiarm star copolymers P(G-co-GEGE-g-EO) with acid-labile polyether core and PEG side chains was synthesized by using the P(G-co-GEGE) copolymers as multifunctional macroinitiators for AROP of EO. The new materials have been characterized in a detailed manner, revealing narrow to moderate molecular weight distributions. The degradation of these polymers under acidic conditions was characterized via SEC and 1H NMR spectroscopy.
Co-reporter:Paul Boehm;Mihail Mondeshki
Macromolecular Rapid Communications 2012 Volume 33( Issue 21) pp:1861-1867
Publication Date(Web):
DOI:10.1002/marc.201200365
Abstract
Block copolymers consisting exclusively of a silicon–oxygen backbone are synthesized by sequential anionic ring-opening polymerization of different cyclic siloxane monomers. After formation of a poly(dimethylsiloxane) (PDMS) block by butyllithium-initiated polymerization of D3, a functional second block is generated by subsequent addition of tetramethyl tetravinyl cyclotetrasiloxane (D4V), resulting in diblock copolymers comprised a simple PDMS block and a functional poly(methylvinylsiloxane) (PMVS) block. Polymers of varying block length ratios were obtained and characterized. The vinyl groups of the second block can be easily modified with a variety of side chains using hydrosilylation chemistry to attach compounds with Si—H bond. Conversion of the hydrosilylation used for polymer modification was investigated.
Co-reporter:Valerie S. Reuss;Mathias Werre
Macromolecular Rapid Communications 2012 Volume 33( Issue 18) pp:1556-1561
Publication Date(Web):
DOI:10.1002/marc.201200307
Abstract
The synthesis of diblock as well as gradient copolymers of N,N-diethyl glycidyl amine (DEGA) with ethylene oxide (EO) via anionic ring-opening polymerization is presented. The polymers exhibit low polydispersities (≤1.13) and molecular weights in the range of 3300–10 200 g mol−1. In PEG-co-PDEGA copolymers, incorporation of 4%–29% DEGA results in tailorable cloud point temperatures in aqueous solution and melting points depending on DEGA content. mPEG-b-PDEGA block copolymers can be quaternized to generate cationic double-hydrophilic polyelectrolyte copolymers with polyether backbone. Furthermore, mPEG-b-PDEGA has been used as dual reducing and capping agent for gold nanoparticle synthesis.
Co-reporter:Sophie S. Müller
Macromolecular Chemistry and Physics 2012 Volume 213( Issue 17) pp:1783-1790
Publication Date(Web):
DOI:10.1002/macp.201200269
Abstract
Synthesis, characterization, and thermal properties of a series of oxetane-functional aliphatic polyesters are investigated. The incorporation of the acid-sensitive 3,3-bis(hydroxymethyl)oxetane (BHMO) into polymers is achieved by using the enzyme CALB (Candida antarctica Lipase B) as a catalyst. This mild synthetic strategy provides well-defined, oxetane-functional polyesters. The enzymatic polycondensation allows for the synthesis of a series of aliphatic polyesters containing various ratios of the difunctional monomers sebacic acid, 1,8-octanediol, and BHMO with molecular weights between 5000–9800 g mol−1 and polydispersity indices (Mw/Mn) in the range of 1.25 and 1.92. Furthermore, crosslinking of BHMO and a polymer sample is carried out via opening of the pending oxetanes.
Co-reporter:Anna M. Fischer;Florian K. Wolf
Macromolecular Chemistry and Physics 2012 Volume 213( Issue 13) pp:1349-1358
Publication Date(Web):
DOI:10.1002/macp.201200082
Abstract
A series of long-chain branched poly(d-/l-lactide)s is synthesized in a two-step protocol by (1) ring-opening polymerization of lactide and (2) subsequent condensation of the preformed AB2 macromonomers promoted by different coupling reagents. The linear AB2 macromonomers are prepared by Sn(Oct)2-catalyzed ROP of D- and L-lactide with 2,2-bis(hydroxymethyl)butyric acid (BHB) as an initiator. Optimization of the polymerization conditions allows for the preparation of well-defined macromonomers (Mw/Mn = 1.09–1.30) with adjustable molecular weights (760–7200 g mol−1). The two-step approach of the synthesis comprises as well the coupling of these AB2 macromonomers and hence allows precise control over the lactide chain length between the branching units in contrast to a random polycondensation.
Co-reporter:Martina Schömer and Holger Frey
Macromolecules 2012 Volume 45(Issue 7) pp:3039-3046
Publication Date(Web):March 28, 2012
DOI:10.1021/ma300249c
Hydrophilic, functional poly(propylene oxide) (PPO) copolymers were prepared by anionic random copolymerization of propylene oxide with the protected glycidyl derivative ethoxy ethyl glycidyl ether (EEGE). The monobenzyl-protected ethylene glycol initiator 2-(benzyloxy)ethanol was used to initiate the polymerization because it allows for the introduction of hydroxyl groups at both ends of the polymer chain. Acidic deprotection permitted selective removal of the acetal protecting groups in the chain or alternatively orthogonal deprotection of the terminal hydroxyl group by catalytic hydrogenation. A series of narrowly distributed hydroxyl-functional PPO copolymers (Mw/Mn < 1.07–1.25 g mol–1) was obtained with varying composition between 2 and 75% glycerol units and molecular weights in the range of 3000 to 8000 g mol–1. Monomer consumption and compositional drift in monomer feed were studied via 1H NMR kinetics, revealing a slightly tapered structure, but confirmed a distribution of EEGE in the polar PPO-based copolymers. Cloud-point measurements showed temperature-dependent water solubility, with LCSTs in the range of 20 to 70 °C. The study demonstrates that an incorporation of 11% of the hydroxyl-functional, linear glycerol units suffices to obtain functional PPO copolymers that are water-soluble at room temperature.
Co-reporter:Christoph Tonhauser, Markus Mazurowski, Matthias Rehahn, Markus Gallei, and Holger Frey
Macromolecules 2012 Volume 45(Issue 8) pp:3409-3418
Publication Date(Web):April 6, 2012
DOI:10.1021/ma3000048
We describe the synthesis of water-soluble diblock and miktoarm star polymers consisting of poly(vinylferrocene) (PVFc) and poly(ethylene oxide) (PEO) blocks. First, end-functionalized poly(vinylferrocene) was generated by end-capping the living carbanionic PVFc chains with benzyl glycidyl ether (BGE) or ethoxy ethyl glycidyl ether (EEGE). Acidic hydrolysis of the EEGE-terminated PVFc partially oxidized the PVFc backbone. However, the dihydroxyl end-functional PVFc was obtained in quantitative yields by hydrogenolysis of the BGE-terminated PVFc. A series of block copolymers and AB2 miktoarm star copolymers was obtained in a second polymerization step, utilizing the respective end-functionalized PVFc as a macroinitiator for the ring-opening polymerization (ROP) of ethylene oxide. All polymers were analyzed in detail, using NMR spectroscopy and size-exclusion chromatography (SEC). Online SEC-viscosimetry as well as MALLS was carried out, confirming the formation of miktoarm structures. Quantitative functionalization and subsequent removal of the acetal and benzyl protective groups, respectively were confirmed by MALDI–ToF mass spectrometry. Molecular weights of the end-functionalized PVFcs range between 1000 and 3600 g mol–1, and block copolymers with 10 000 to 50 000 g mol–1 overall molar masses were synthesized. In addition, the water-soluble block copolymers were investigated by differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA). For characterization of the morphology in aqueous solution, transmission electron microscopy (TEM) was performed, showing micelles and multicompartment micellar structures.
Co-reporter:Christoph Schüll, Lutz Nuhn, Christine Mangold, Eva Christ, Rudolf Zentel, and Holger Frey
Macromolecules 2012 Volume 45(Issue 15) pp:5901-5910
Publication Date(Web):July 18, 2012
DOI:10.1021/ma300972v
The synthesis of hyperbranched polyglycerol dendron analogues with precisely one focal amino functionality (H2N-hbPG) and their use for the synthesis of linear-hyperbranched graft-copolymers in a grafting-to approach is reported. By use of N,N-dibenzyl tris(hydroxylmethyl) aminomethane as a novel initiator for the ring-opening multibranching polymerization of glycidol, dendron analogues with one focal amino functionality of molecular weights ranging from 500 to 15000 g mol–1 and narrow to moderate polydispersities (Mw/Mn = 1.2–1.9) were synthesized, as confirmed by NMR and SEC. After removal of the benzyl protective groups, the accessibility and selective transformation of the focal amino group was demonstrated by NMR and MALDI–ToF MS. H2N-hbPG was then selectively grafted to a linear reactive ester polymer backbone, poly(pentafluorophenol methacrylate) (PPFPMA), to obtain linear polymers with highly branched side chains of high molecular weights (Mn > 126 kg mol–1) and low polydispersities (Mw/Mn = 1.1–1.3), as shown by SEC. Successful attachment of the hyperbranched polyether structure to the linear backbone was confirmed by 19F NMR and fluorescence spectroscopy. The modular approach introduced bears some analogy to the convergent dendrimer synthesis and describes the first linear-hyperbranched graft-copolymers prepared in a grafting-to approach, resulting in hyperbranched brush-type polymers with the highest molecular weight reported to date.
Co-reporter:Valerie S. Reuss, Boris Obermeier, Carsten Dingels, and Holger Frey
Macromolecules 2012 Volume 45(Issue 11) pp:4581-4589
Publication Date(Web):May 21, 2012
DOI:10.1021/ma300292m
The first application of N,N-diallylglycidylamine (DAGA) as a monomer for anionic ring-opening polymerization is presented. The monomer is obtained in a one-step procedure using epichlorohydrin and N,N-diallylamine. Both random and block copolymers consisting of poly(ethylene glycol) and poly(N,N-diallylglycidylamine) with adjusted DAGA ratios from 2.5 to 24% have been prepared, yielding well-defined materials with low polydispersities (Mw/Mn) in the range 1.04–1.19. Molecular weights ranged between 2600 and 10 300 g mol–1. Isomerization of allylamine to enamine structures during polymerization depending on time, temperature, and counterion has been realized. The kinetics of the formation of the copolymer structure obtained by random copolymerization was investigated, using time-resolved 1H NMR measurements and 13C NMR triad sequence analysis. A tapered character of the monomer incorporation was revealed in the course of the concurrent copolymerization of EO and DAGA. The thermal behavior of the copolymers in both bulk and aqueous solution has been studied, revealing LCSTs in the range 29–94 °C. Quantitative removal of protective groups via double-bond isomerization mediated by Wilkinson’s catalyst and subsequent acidic hydrolysis yielded multiamino-functional PEG copolymers with tapered or block structure. Accessibility of liberated primary amines for further transformation was demonstrated in a model reaction by derivatization with acetic anhydride. In contrast to previous approaches, the DAGA monomer permits the synthesis of block copolymers with PEG block combined with multiamino-functional polyether block.
Co-reporter:Christoph Tonhauser, Adrian Natalello, Holger Löwe, and Holger Frey
Macromolecules 2012 Volume 45(Issue 24) pp:9551-9570
Publication Date(Web):November 30, 2012
DOI:10.1021/ma301671x
Microflow technology, i.e., the use of microfluidic devices for continuous flow synthesis, represents a highly useful and increasingly popular method in organic chemistry. Recently, also an increasing number of polymer synthesis protocols attain benefit from this technique. In particular, the control of highly exothermic, fast polymerization reactions can be improved due to the excellent heat and mass transfer within the small dimensions of the microreactors. Continuous flow setups with different micromixer geometries and flow patterns are currently used for the preparation of a variety of macromolecular architectures by ionic and (controlled) radical polymerization techniques. This Perspectives reviews recent developments in synthetic strategies and reactor design for the homogeneous synthesis of polymers in microflow systems and emphasizes future challenges and promise for applications. Polymer synthesis by radical, anionic, cationic, and coordinative polymerization is considered as well as different polymer topologies generated (linear, branched, and dendritic architectures).
Co-reporter:Carsten Dingels;Dr. Frederik Wurm;Dr. Manfred Wagner;Dr. Harm-Anton Klok;Dr. Holger Frey
Chemistry - A European Journal 2012 Volume 18( Issue 52) pp:16828-16835
Publication Date(Web):
DOI:10.1002/chem.201200182
Abstract
The covalent attachment of poly(ethylene glycol) (PEG) to therapeutically active proteins (PEGylation) has become an important method to deal with the pharmacological difficulties of these polypeptides, such as short body-residence times and immunogenicity. However, the derivatives of PEG used for PEGylation lack further functional groups that would allow the addition of targeting or labeling moieties. Squaric acid diethyl ester was used for the chemoselective single-step activation of poly(ethylene glycol)s into the respective ester amides. The resultant selective protein-reactive poly(ethylene glycol)s were investigated with respect to their selectivity towards amino acid residues in bovine serum albumin (as a model protein). The presented procedure relies on a robust two-step protocol and was found to be selective towards lysine residues; the activated polyethers are efficient and stoichiometric PEGylation agents with a remarkable hydrolytic stability over a period of several days. By adjusting the pD value of the conjugation mixture, the chemoselectivity of the activated PEGs towards the α- and ε-amino groups of lysine methyl ester was effectively changed.
Co-reporter:Frederik Wurm, Holger Frey
Progress in Polymer Science 2011 Volume 36(Issue 1) pp:1-52
Publication Date(Web):January 2011
DOI:10.1016/j.progpolymsci.2010.07.009
Concurrent with the rapid development of both dendrimers and hyperbranched polymers, a novel class of block copolymer architectures has emerged from the combination of these dendritic architectures with linear chains, the “linear–dendritic block copolymers” (LDBCs). This review gives a comprehensive summary of the state of the art in this rapidly developing field from pioneering early work to promising recent approaches.The different strategies leading to these hybrid architectures with either perfect dendrimer/dendron building blocks or imperfect, yet more conveniently accessible hyperbranched segments, are reviewed and compared. The consequences of the unusual polymer topology for supramolecular structures both in solution and in the solid state are summarized, and important differences in comparison with classical linear block copolymer structures are highlighted. Current challenges in the area of block copolymers, nanotechnology and potential applications of linear–dendritic block copolymers are also considered.
Co-reporter:Sung-Il Lee, Martina Schömer, Huagen Peng, Kirt A. Page, Daniel Wilms, Holger Frey, Christopher L. Soles, and Do Y. Yoon
Chemistry of Materials 2011 Volume 23(Issue 11) pp:2685
Publication Date(Web):May 17, 2011
DOI:10.1021/cm103696g
Co-reporter:Christoph Tonhauser, Boris Obermeier, Christine Mangold, Holger Löwe and Holger Frey
Chemical Communications 2011 vol. 47(Issue 31) pp:8964-8966
Publication Date(Web):07 Jul 2011
DOI:10.1039/C1CC12956B
A series of block copolymers bearing a single amino in-chain functionality was synthesized viaanionic polymerization of styrene and ethylene oxide. By means of both a conventional and a continuous setup, living polystyrene was quantitatively end functionalized with an oxirane (DBAG) prior to the polymerization of the poly(ethylene oxide) segment. The in-chain amine was conjugated with a fluorescent dye.
Co-reporter:Boris Obermeier and Holger Frey
Bioconjugate Chemistry 2011 Volume 22(Issue 3) pp:436
Publication Date(Web):February 14, 2011
DOI:10.1021/bc1004747
A series of random copolymers comprising ethylene oxide (EO) and 0−100% allyl glycidyl ether (AGE) has been prepared by anionic ring-opening polymerization with molecular weights between 5000 and 13 600 g/mol and polydispersity indices in the range of 1.04−1.19. As key for the homogeneity of the PEG conjugates, real-time 1H NMR polymerization kinetics, 13C NMR analysis of triad sequence distribution, and analysis of the thermal behavior by differential scanning calorimetry (DSC) revealed a distinctive random copolymer structure. Via thiol−ene coupling (TEC), showing mainly “click” characteristics and nearly quantitative yields, PEG derivatives with multiple amino, carboxy, or hydroxy functionalities have been prepared, providing suitable reactivities for further attachment. Without further modification, P(EO-co-AGE)s were conjugated with cysteine or the tripeptide glutathione (GSH) via TEC, resulting in well-defined hybrid materials with multiple peptide units conjugated to the PEG backbone. The results demonstrate superior loading capacity of the copolymers in comparison to the PEG homopolymer.
Co-reporter:Christine Mangold;Boris Obermeier;Frederik Wurm
Macromolecular Rapid Communications 2011 Volume 32( Issue 23) pp:1930-1934
Publication Date(Web):
DOI:10.1002/marc.201100489
Abstract
The lower critical solution temperature (LCST) behavior of novel poly(ethylene glycol) (PEG)-based copolymers bearing multiple functional groups, obtained by anionic ring-opening (co)polymerization (AROP), has been investigated. Variable comonomer ratios of ethylene oxide (EO) and the corresponding oxiranes isopropylidene glyceryl glycidyl ether (IGG), ethoxyl vinyl glycidyl ether (EVGE), allyl glycidyl ether (AGE), or N,N-dibenzyl amino glycidyl (DBAG), particularly designed to implement functional groups at the PEG backbone, were found to influence the LCST behavior. Sharp transitions from translucent to opaque solutions, comparable to other well-established stimuli-responsive polymers, were observed at temperatures ranging from 9 to 82 °C. The influence of the side group hydrophobicity could be quantified by the comparison of the different copolymer systems observed.
Co-reporter:Anja Thomas;Florian K. Wolf
Macromolecular Rapid Communications 2011 Volume 32( Issue 23) pp:1910-1915
Publication Date(Web):
DOI:10.1002/marc.201100432
Abstract
Linear, protected ω-methoxy oligo(glycerol) methacrylate (OGlyPMA) macromonomers are synthesized via anionic ring-opening polymerization of ethoxyethyl glycidyl ether (EEGE) followed by termination with methacrylic acid anhydride ( = 3–11, PDI < 1.30). The covalently bound methacrylate moiety allows the homopolymerization of OGlyPMA as well as copolymerization with low molecular weight comonomers. In homopolymerizations, macromonomers are polymerized by atom transfer radical polymerization (ATRP) yielding well-defined graft polymers ( = 20 000–30 000 g mol−1). Acidic hydrolysis of the protecting groups releases water-soluble polyhydroxy-functional structures. First results on the copolymerization with 2-hydroxyethyl methacrylate (HEMA) are given in the final part of this work.
Co-reporter:Martina Schömer
Macromolecular Chemistry and Physics 2011 Volume 212( Issue 22) pp:2478-2486
Publication Date(Web):
DOI:10.1002/macp.201100386
Abstract
Multiarm star copolymers consisting of the polyether-polyol hyperbranched poly(ethylene glycol) (hbPEG) as core and poly(L-lactide) (PLLA) arms are synthesized via the organobase- catalyzed ring-opening polymerization of lactide using hbPEG as a multifunctional macroinitiator. Star copolymers with high molecular weights up to 792 000 g mol−1 are prepared. Detailed 2D NMR analysis provides evidence for the attachment of the PLLA arms to the core and reveals that the adjustment of the monomer/initiator ratio enables control of the arm length. Size exclusion chromatography measurements show narrow molecular weight distributions. Thermal analysis reveals a lower glass transition temperature, melting point, and degree of crystallization for the star-shaped polylactides compared to linear polylactide.
Co-reporter:Carsten Dingels;Martina Schömer ; Dr. Holger Frey
Chemie in unserer Zeit 2011 Volume 45( Issue 5) pp:338-349
Publication Date(Web):
DOI:10.1002/ciuz.201100551
Abstract
Poly(ethylenglykol) ist in unserem Leben allgegenwärtig. Wir begegnen diesem biokompatiblen und hervorragend wasserlöslichen Polymer, meist ohne dass uns dies bewusst ist, in fast allen Bereichen des alltäglichen Bedarfs: Von Haut- und Haarpflegeartikeln, über Kosmetik- und Styling-Produkten bis hin zu Lebensmitteln und Medikamenten. Selbst in der maritimen Militärtechnologie und bei der Konservierung geborgener Kulturgüter wird es eingesetzt. Dieser Artikel beschäftigt sich mit der Darstellung, den teils überraschenden Eigenschaften und der Anwendung dieses strukturell simplen, aber faszinierend vielseitigen Polymers.
Today, a life without poly(ethylene glycol) is hardly imaginable. One encounters PEG in nearly every aspect of our daily needs: from cosmetics to hairstyling products and from medicines to food. Even in maritime military technology and the preservation of recovered cultural possessions PEG can be found. This article deals with the synthesis, the surprising properties, and the application of this structurally simple yet fascinating and important polymer.
Co-reporter:Dr. Boris Obermeier;Dr. Frederik Wurm;Dipl.-Chem. Christine Mangold;Dr. Holger Frey
Angewandte Chemie 2011 Volume 123( Issue 35) pp:8136-8146
Publication Date(Web):
DOI:10.1002/ange.201100027
Abstract
Maßgeschneiderte makromolekulare Strukturen stellen auf dem sich stetig entwickelnden Feld der Polymertherapeutika die Schlüsselkomponente dar, um das Potential bioaktiver Konjugate auszuschöpfen. Die jüngsten Entwicklungen auf dem Gebiet der Wirkstoffkonjugate zur Krebsbehandlung zeigen, dass multifunktionelle Polymere als Wirkstoffträger immer bedeutender werden. Die Anwendung des vermeintlich am besten geeigneten Polymers, Poly(ethylenglycol) (PEG), ist hier jedoch durch die limitierte Zahl funktioneller Gruppen eingeschränkt. Als Resultat ziehen multifunktionelle Copolymere basierend auf Ethylenoxid (EO) und einem entsprechenden Epoxid als Comonomer (mf-PEGs) zunehmend Aufmerksamkeit auf sich. Wohldefiniert dargestellt durch lebende anionische Polymerisation in Kombination mit modernen Charakterisierungsmethoden – beispielsweise Messungen der Polymerisationskinetik über 1H-NMR-Spektroskopie in Echtzeit –, repräsentiert diese aufkommende Klasse an Polymeren eine leistungsstarke Plattform zur Synthese von Bio- und Wirkstoffkonjugaten.
Co-reporter:Boris Obermeier, Peter Langguth, and Holger Frey
Biomacromolecules 2011 Volume 12(Issue 2) pp:
Publication Date(Web):December 30, 2010
DOI:10.1021/bm1012037
Partially quarternized poly(methacrylate) terpolymers (Q-BBMCs) have been synthesized, based on the basic butylated methacrylate copolymer (BBMC/EUDRAGIT E), an excipient approved by the Food and Drug Administration (FDA) and to date mainly applied for tablet coatings. Via straightforward polymer modification reactions, a series of Q-BBMCs with quarternization degrees of 22%, 42%, and 65% has been prepared. Apical to basolateral transport across Caco-2 cell monolayers was investigated, employing the paracellular transported compounds trospium and mannitol. At pH 6.5 quarternization resulted in increased permeation enhancement up to 2.8-fold compared to BBMC, that is, up to 7.3-fold compared to control. Moreover, measurements of the transepithelial electrical resistance (TEER) revealed a special advantage of the quarternized poly(methacrylate) terpolymers with respect to the pH range, in which the polymers exhibit biological activity as permeation enhancers. Whereas at pH 6.5 TEER dropped within 30 min below 30% of the initial value for all polymers, at pH 7.4 this effect solely occurred for Q-BBMCs, meaning a significant extension of the pH range relevant for drug permeation. In a subsequent period of 6 h, also excellent recovery was observed.
Co-reporter:Dr. Boris Obermeier;Dr. Frederik Wurm;Dipl.-Chem. Christine Mangold;Dr. Holger Frey
Angewandte Chemie International Edition 2011 Volume 50( Issue 35) pp:7988-7997
Publication Date(Web):
DOI:10.1002/anie.201100027
Abstract
In the rapidly evolving multidisciplinary field of polymer therapeutics, tailored polymer structures represent the key constituent to explore and harvest the potential of bioactive macromolecular hybrid structures. In light of the recent developments for anticancer drug conjugates, multifunctional polymers are becoming ever more relevant as drug carriers. However, the potentially best suited polymer, poly(ethylene glycol) (PEG), is unfavorable owing to its limited functionality. Therefore, multifunctional linear copolymers (mf-PEGs) based on ethylene oxide (EO) and appropriate epoxide comonomers are attracting increased attention. Precisely engineered via living anionic polymerization and defined with state-of-the-art characterization techniques—for example real-time 1H NMR spectroscopy monitoring of the EO polymerization kinetics—this emerging class of polymers embodies a powerful platform for bio- and drug conjugation.
Co-reporter:Anna Maria Hofmann, Robert Wipf, Bernd Stühn, and Holger Frey
Macromolecules 2011 Volume 44(Issue 17) pp:6767-6775
Publication Date(Web):August 12, 2011
DOI:10.1021/ma201210r
Synthesis, thermal properties, and the liquid crystalline (LC) order of polymers consisting of a single mesogenic cholesterol unit and flexible, linear polyglycerol (PG) or poly(glyceryl glycerol) (PGG) chains have been investigated. Incorporation of the single mesogen has been achieved by using cholesterol directly as an initiator for the oxyanionic ring-opening polymerization (ROP) of ethoxyethyl glycidyl ether (EEGE) or isopropylidene glyceryl glycidyl ether (IGG). The controlled polymerization allowed the synthesis of a series of peculiar rod–coil type polyethers with molecular weights of 600–2300 g/mol, representing a degree of polymerization (DPn) of 4–30 for both PG and PGG with the polydispersity Mw/Mn in the range of 1.07–1.25. The resulting linear PGs exhibit extremely stable thermotropic LC order in a broad temperature range up to 260 °C, forming mainly layered smectic A (SmA) phases with varying layer thicknesses, depending on the degree of polymerization of the respective polymer structure. LC phases were observed up to a chain length of 26 glycerol units, while PGGs showed no LC order. This is explained both by the steric hindrance of the branched monomer units and the higher hydrophilicity of the polymer backbone. Permethylation of the cholesterol-PG samples resulted in strongly reduced LC order or in the entire loss of the self-assembly in LC phases, which is a consequence of the disappearance of hydrogen-bonding between the functional coil segments. Detailed characterization of the phase behavior of the polymers has been achieved by differential scanning calorimetry (DSC), polarized optical microscopy (POM), and small-angle X-ray scattering (SAXS), confirming the smectic layer structure of the materials.
Co-reporter:Christine Mangold, Carsten Dingels, Boris Obermeier, Holger Frey, and Frederik Wurm
Macromolecules 2011 Volume 44(Issue 16) pp:6326-6334
Publication Date(Web):July 22, 2011
DOI:10.1021/ma200898n
Introduction of highly reactive vinyl ether moieties along a poly(ethylene glycol) (PEG) backbone has been realized by copolymerization of the novel epoxide monomer ethoxy vinyl glycidyl ether (EVGE) with ethylene oxide (EO). A series of copolymers with varying structure (block and random) as well as EVGE comonomer content (5–100%) with molecular weights in the range of 3,900–13,200 g/mol and narrow molecular weight distributions (Mw/Mn = 1.06–1.20) has been synthesized and characterized with respect to their microstructure and thermal properties. The facile transformation of the vinyl ether side chains in click type reactions was verified by two different post polymerization modification reactions: (i) thiol–ene addition and (ii) acetal formation, employing various model compounds. Both strategies are very efficient, resulting in quantitative conversion. The rapid and complete acetal formation with alcohols results in an acid-labile bond and is thus highly interesting with respect to biomedical applications that require slow or controlled release of a drug, while the thiol–ene addition to a vinyl ether prevents cross-linking efficiently compared to other double bonds.
Co-reporter:Anna Maria Hofmann, Frederik Wurm, and Holger Frey
Macromolecules 2011 Volume 44(Issue 12) pp:4648-4657
Publication Date(Web):May 18, 2011
DOI:10.1021/ma200367c
Polymer-coated liposomes, particularly poly(ethylene glycol) (PEG)-substituted liposomes, have emerged as long-circulating carrier systems for drug delivery and diagnostic purposes. A rapid synthesis of three different types of multifunctional lipids with structurally diverse hydrophilic, polyether-based architectures via one- or two-pot approaches is described. Architectural variation is achieved by the combination of different oxyanionic polymerization strategies and various glycidyl ether building units. Branched polyglycerol lipids have been prepared via cholesterol- or 1,2-bis-n-alkyl glyceryl ether-initiated, oxyanionic ring-opening polymerization (ROP) of protected glycidyl ethers and glycidol, respectively. In addition to these polyglycerol-based lipids, we describe the synthesis of multifunctional PEGs as the hydrophilic part of the lipid, which can be compared to conventional stealth lipids, but bear an adjustable number of hydroxyl functions within the PEG backbone. These lipids can be readily obtained by random copolymerization of ethylene oxide and protected glycidyl ethers, such as ethoxyethyl glycidyl ether (EEGE) and isopropylidene glyceryl glycidyl ether (IGG). Polydispersities Mw/Mn of the amphiphilic polyether structures were in the range of 1.04–1.2 for the linear structures and 1.1–1.6 for the hyperbranched lipids. Critical micelle concentrations (CMC) have been determined via the pyrene fluorescence method and were in the range of 1.4–40.7 mg/L, correlated to molecular weight and functionality of the polar polyether segment. Liposomes containing these hydroxy-functional lipids have been prepared via the membrane extrusion method and have been visualized by transmission electron microscopy (TEM) and cryo-TEM.
Co-reporter:Adrian Natalello, Christoph Tonhauser, Elena Berger-Nicoletti, and Holger Frey
Macromolecules 2011 Volume 44(Issue 24) pp:9887-9890
Publication Date(Web):November 30, 2011
DOI:10.1021/ma2023793
Co-reporter:Daniel Wilms, Salah-Eddine Stiriba, and Holger Frey
Accounts of Chemical Research 2010 Volume 43(Issue 1) pp:129
Publication Date(Web):September 28, 2009
DOI:10.1021/ar900158p
Dendritic macromolecules with random branch-on-branch topology, termed hyperbranched polymers in the late 1980s, have a decided advantage over symmetrical dendrimers by virtue of typically being accessible in a one-step synthesis. Saving this synthetic effort once had an unfortunate consequence, though: hyperbranching polymerization used to result in a broad distribution of molecular weights (that is, very high polydispersities, often Mw/Mn > 5). By contrast, a typical dendrimer synthesis yields a single molecule (in other words, Mw/Mn = 1.0), albeit by a labor-intensive, multistep process. But 10 years ago, Sunder and colleagues reported the controlled synthesis of well-defined hyperbranched polyglycerol (PG) via ring-opening multibranching polymerization (ROMBP) of glycidol. Since then, hyperbranched and polyfunctional polyethers with controlled molar mass and low polydispersities (Mw/Mn = 1.2−1.9) have been prepared, through various monomer addition protocols, by ROMBP. In this Account, we review the progress in the preparation and application of these uniquely versatile polyether polyols over the past decade.Hyperbranched PGs combine several remarkable features, including a highly flexible aliphatic polyether backbone, multiple hydrophilic groups, and excellent biocompatibility. Within the past decade, intense efforts have been directed at the optimization of synthetic procedures affording PG homo- and copolymers with different molecular weight characteristics and topology. Fundamental parameters of hyperbranched polymers include molar mass, polydispersity, degree of branching, and end-group functionality. Selected approaches for optimizing and tailoring these characteristics are presented and classified with respect to their application potential. Specific functionalization in the core and at the periphery of hyperbranched PG has been pursued to meet the growing demand for novel specialty materials in academia and industry.A variety of fascinating synthetic approaches now provide access to well-defined, complex macromolecular architectures based on polyether polyols with low polydispersity. For instance, a variety of linear−hyperbranched block copolymers has been reported. The inherent attributes of PG-based materials are useful for a number of individual implementation concepts, such as drug encapsulation or surface modification. The excellent biocompatibility of PG has also led to rapidly growing significance in biomedical applications, for example, bioconjugation with peptides, as well as surface attachment for the creation of protein-resistant surfaces.
Co-reporter:Christine Mangold;Frederik Wurm;Boris Obermeier
Macromolecular Rapid Communications 2010 Volume 31( Issue 3) pp:258-264
Publication Date(Web):
DOI:10.1002/marc.200900472
Co-reporter:Christine Mangold;Frederik Wurm;Boris Obermeier
Macromolecular Rapid Communications 2010 Volume 31( Issue 3) pp:
Publication Date(Web):
DOI:10.1002/marc.201090004
Co-reporter:Christoph Tonhauser
Macromolecular Rapid Communications 2010 Volume 31( Issue 22) pp:1938-1947
Publication Date(Web):
DOI:10.1002/marc.201000353
Co-reporter:Christoph Tonhauser;Daniel Wilms;Yasmin Korth;Christian Friedrich
Macromolecular Rapid Communications 2010 Volume 31( Issue 24) pp:2127-2132
Publication Date(Web):
DOI:10.1002/marc.201000473
Co-reporter:Daniel Wilms;Martina Schömer;Frederik Wurm;M. Iris Hermanns;C. James Kirkpatrick
Macromolecular Rapid Communications 2010 Volume 31( Issue 20) pp:1811-1815
Publication Date(Web):
DOI:10.1002/marc.201000329
Co-reporter:Maria Doycheva;Elena Berger-Nicoletti;Frederik Wurm
Macromolecular Chemistry and Physics 2010 Volume 211( Issue 1) pp:35-44
Publication Date(Web):
DOI:10.1002/macp.200900377
Co-reporter:Frederik Wurm;Anna Maria Hofmann;Anja Thomas;Carsten Dingels
Macromolecular Chemistry and Physics 2010 Volume 211( Issue 8) pp:932-939
Publication Date(Web):
DOI:10.1002/macp.200900652
Co-reporter:Anna M. Fischer and Holger Frey
Macromolecules 2010 Volume 43(Issue 20) pp:8539-8548
Publication Date(Web):September 22, 2010
DOI:10.1021/ma101710t
A series of (hyper)branched poly(glycolide) copolymers has been prepared by copolymerization of glycolide (GA) with 2,2-bis(hydroxymethyl)butyric acid (BHB) via combined ROP/AB2-polycondensation. Polymerization was conducted in bulk and catalyzed by stanneous-2-ethyl hexanoate (Sn(Oct)2). The branched topology of the resulting polyesters was studied in detail by 1D- and 2D-NMR spectroscopy and confirmed by the synthesis and characterization of model compounds. The AB2 monomer BHB was incorporated either as a dendritic or focal unit, but hardly in linear or terminal mode. As expected for multifunctional polycondensation, SEC measurements showed polydisperse products with polydispersity index in the range of 1.88 to 3.40. Mn of the copolymers varied from 1100 to 4000 g/mol. MALDI−TOF MS analysis allowed to verify the main polymeric species. Furthermore, MALDI−TOF evidenced incorporation of several BHB units per macromolecule, confirming a successful condensation reaction and the formation of branched copolymers. Detailed 1H NMR characterization (1D and 2D methods) permitted calculation of the molar composition, the conversion and the degree of branching (DB), which ranged between 0.12 and 0.44. Differential scanning calorimetry (DSC) measurements showed that in contrast to linear PGA (Tm > 220 °C) the melting behavior and the glass-transition temperature of the branched poly(glycolide) copolymers changed drastically. The presence of dendritic units in the polymer backbone resulted in a depression of the melting point and amorphous materials at amounts of BHB exceeding 15%. The amorphous hyperbranched poly(glycolide) copolymers show enhanced solubility in common solvents (e.g., acetone, ethyl acetate, THF) and improved processability in contrast to linear PGA and possess potential for use in slow or controlled drug release systems.
Co-reporter:Christine Mangold, Frederik Wurm, Boris Obermeier, and Holger Frey
Macromolecules 2010 Volume 43(Issue 20) pp:8511-8518
Publication Date(Web):September 29, 2010
DOI:10.1021/ma1015352
A series of poly(ethylene glycol-co-isopropylidene glyceryl glycidyl ether) (P(EO-co-IGG)) random copolymers with different fractions of 1,2-isopropylidene glyceryl glycidyl ether (IGG) units was synthesized. After acidic hydrolysis a new type of “functional PEGs”, namely poly(ethylene glycol-co-glyceryl glycerol) (P(EO-co-GG)) was obtained. Using an initiator that releases a terminal amino moiety after deprotection, functional end groups with orthogonal reactivity to the in-chain groups were obtained. All polymers showed narrow molecular weight distributions (1.07−1.19), and control of the molecular weights was achieved in the range 5000−30 000 g/mol. Random incorporation of both comonomers was verified by monitoring the copolymerization kinetics via real-time 1H NMR spectroscopy during the polymerization and by characterization of the triad sequence distribution, relying on 13C NMR analysis. Using the 1,2-diol component of the side chains allows for attachment and facile acid-catalyzed release of molecules bearing ketone/aldehyde functionalities. This renders the materials potentially useful as support for reagents, drugs or catalysts. This was demonstrated using benzaldehyde as a model compound. DSC was carried out on all samples, showing amorphous structures upon incorporation of IGG fractions exceeding 15%.
Co-reporter:Valerie S. Reuss and Holger Frey
Macromolecules 2010 Volume 43(Issue 20) pp:8462-8467
Publication Date(Web):September 24, 2010
DOI:10.1021/ma1016715
A new acetal-protected monomer for Wurtz-type coupling to polysilanes, dichloro(3-(2,2-dimethyl-1,3-dioxolane-4-yloxy)propyl)methylsilane, referred to as dichloro(isopropylidene glyceryl propyl ether)methylsilane (DCIMS), has been introduced to synthesize a series of protected linear polysilane copolymers, poly[di-n-hexylsilane-co-(isopropylidene glyceryl propyl ether)methylsilane] (P(DHS-co-IMS)) via alkali-mediated reductive Wurtz-type coupling. The acetal protecting group proved stable under the harsh polymerization conditions. Differential scanning calorimetry combined with 1H, 13C, and 29Si NMR measurements confirmed composition and random structure of the obtained copolymers. After separation of the cylic fraction, this route yielded defined linear polysilane copolymers with monomodal molecular weight distributions (2000−98700 g mol−1 (SEC)) and polydispersities in the range 1.61−2.60. Subsequent cleavage of the acetal protecting groups under acidic conditions resulted in the multihydroxy-functional polysilanes poly[di-n-hexylsilane-co-(glyceryl propyl ether)methylsilane] (P(DHS-co-GMS)).
Co-reporter:Lourdes Pastor-Pérez, Ulrike Kemmer-Jonas, Frederik Wurm, Salah-Eddine Stiriba, Julia Pérez-Prieto, and Holger Frey
Macromolecules 2010 Volume 43(Issue 23) pp:9583-9587
Publication Date(Web):November 10, 2010
DOI:10.1021/ma101660s
Co-reporter:Anna Maria Hofmann, Frederik Wurm, Eva Hühn, Thomas Nawroth, Peter Langguth and Holger Frey
Biomacromolecules 2010 Volume 11(Issue 3) pp:
Publication Date(Web):February 1, 2010
DOI:10.1021/bm901123j
We describe the synthesis of linear-hyperbranched lipids for liposome preparation based on linear poly(ethylene glycol) (PEG) and hyperbranched polyglycerol (PG). Molecular weights were adjusted to values around 3000 g/mol with varying degrees of polymerization of the linear and the branched segments in analogy to PEG-based stealth lipids; polydispersities were generally low and below 1.3. The hydrophobic anchors were introduced into the lipid structures as initiators for the anionic polymerization of ethylene oxide and are either based on cholesterol or on different aliphatic glyceryl ethers. Complete incorporation of the apolar initiators was evidenced by MALDI-ToF analysis at all stages of the reaction. The linear-hyperbranched polyether lipid is incorporated as the polyfunctional shell in liposome formulations together with 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC). The resulting liposomes were subsequently characterized via dynamic light scattering (DLS) and small angle neutron scattering (SANS) as well as transmission electron microscopy (TEM), demonstrating the formation of unilamellar liposomes in the size range of 40 to 50 nm.
Co-reporter:Christoph Tonhauser, Daniel Wilms, Frederik Wurm, Elena Berger Nicoletti, Michael Maskos, Holger Löwe and Holger Frey
Macromolecules 2010 Volume 43(Issue 13) pp:5582-5588
Publication Date(Web):May 28, 2010
DOI:10.1021/ma902849r
We describe the synthesis of end-functionalized polystyrenes by living anionic polymerization in a microstructured reactor via termination by acetal-protected functional epoxides. Initiation of styrene polymerization by alkyllithium takes place in a micromixing device with efficient heat and mass transfer properties. A newly developed continuous polymerization−termination sequence enabled quantitative functionalization of the living carbanions by nucleophilic displacement with different, specifically designed glycidyl ethers (ethoxy ethyl glycidyl ether (EEGE), 1,2-isopropylidene glyceryl glycidyl ether (IGG), and trans-2-phenyl-1,3-dioxane glycidyl ether (PDGE)). Upon acidic hydrolysis the end-capped polystyrenes release multiple hydroxyl groups (2−3) at the chain end. Temperature and flow rates have been varied to control molecular weights and to optimize the reaction conditions for maximum polymerization and termination efficiency. The polymers were analyzed in detail using NMR spectroscopy, size exclusion chromatography (SEC), and MALDI-ToF-MS. Molecular weights of the samples prepared ranged between 1800 and 9000 g/mol. For all of the novel termination agents full termination was confirmed by MALDI-ToF MS. The approach presented is applicable for a large variety of monomers that are polymerizable by carbanionic polymerization.
Co-reporter:Boris Obermeier, Frederik Wurm and Holger Frey
Macromolecules 2010 Volume 43(Issue 5) pp:2244-2251
Publication Date(Web):February 15, 2010
DOI:10.1021/ma902245d
The synthesis of poly(ethylene glycol) (PEG) copolymers with multiple amino functionalities within the chain is described, relying on an epoxide comonomer bearing a protected amino group. N,N-dibenzyl amino glycidol (DBAG) and ethylene oxide (EO) were copolymerized via anionic polymerization, leading to well-defined polymers with varied comonomer content and low polydispersities (Mw/Mn in the range of 1.1 to 1.2). Subsequent hydrogenolysis with Pearlman’s catalyst afforded poly(ethylene glycol-co-amino glycerol)s (PEG-co-PAG) with a precisely adjusted number of randomly incorporated amino groups in the range of 2−15%. For the first time, the kinetics of an EO copolymerizations have has been directly monitored by 1H NMR spectroscopy in real time. Monomer consumption and compositional drift in monomer feed have been studied for various reaction temperatures, revealing a slightly tapered yet random DBAG distribution in the copolymers. The random structure of the copolymers was confirmed by detailed 13C NMR characterization of EO- and DBAG-centered triad sequence distribution and DSC measurements.
Co-reporter:Florian K. Wolf, Anna M. Hofmann and Holger Frey
Macromolecules 2010 Volume 43(Issue 7) pp:3314-3324
Publication Date(Web):March 19, 2010
DOI:10.1021/ma902844m
On the basis of a new acetal-protected glycerol monomethacrylate monomer (cis-1,3-benzylidene glycerol methacrylate/BGMA) a series of potentially biocompatible and partially biodegradable homo- and block copolymers were synthesized. ATRP polymerization of BGMA yielded well-defined polyacrylates with pendant benzylidene acetal groups and high glass transition temperatures (115−130 °C). This hydrophobic poly(cis-1,3-benzylidene glycerol methacrylate) could be readily transformed into the hydrophilic and water-soluble poly(1,3-dihydroxypropyl methacrylate), referred to as poly(isoglycerol methacrylate) (PIGMA). It exclusively contains primary hydroxyl groups and therefore differs significantly from the commonly known poly(glycerol methacrylate) (PGMA). Block copolymer systems based on poly(lactide) and BGMA were realized via two orthogonal living polymerization techniques starting from a bifunctional initiator, employing first atom transfer radical polymerization (ATRP) of BGMA and in the second step organo-base catalyzed polymerization of l- or d-lactide. This route provides well-defined block copolymers of low polydispersity (PDI 1.12−1.17) and molecular weights in the range of 7000 to 30 000 g/mol (NMR). Rapid and highly selective acetal hydrolysis of the PBGMA block resulted in the release of the hydrophilic and water-soluble poly(1,3-dihydroxypropyl methacrylate) (poly(isoglycerol methacrylate), PIGMA). Acidic hydrolysis of the acetal protecting groups of poly(BGMA)-b-poly(lactide) copolymers proceeded smoothly to amphiphilic structures, notably without affecting the potentially labile polyester block. The novel PIGMA-b-PLLA copolymers are capable of supramolecular self-assembly to spherical aggregate structures in aqueous environment. The polymers generally exhibited low aggregation constants (CAC: 8−20 mg/L). Because of the unique feature of stereocomplex formation of poly(lactide), the corresponding aggregate morphology could be adjusted by mixing two nearly identical PIGMA-b-PLA copolymers with enantiomeric poly(lactide blocks) in a 1:1 ratio. In this case the uniformly shaped micelles (20 nm) changed to large vesicles with diameters ranging from 600 to 1400 nm. These features render this new type of amphiphilic block copolymers promising for drug delivery applications.
Co-reporter:Frederik Wurm ; Johannes Klos ; Hans Joachim Räder
Journal of the American Chemical Society 2009 Volume 131(Issue 23) pp:7954-7955
Publication Date(Web):May 22, 2009
DOI:10.1021/ja9019148
Linear-hyperbranched, heterobifunctional α,ωn telechelic block copolymers consisting of a linear poly(ethylene glycol) (PEG) chain and a hyperbranched polyglycerol (PG) block have been prepared in five steps, using a protected amino-functional initiator. The polyfunctionality ωn (OH groups) can be adjusted by the degree of polymerization (DPn) of the polyglycerol block. Subsequent introduction of a single biotin unit by amidation in α-position permitted noncovalent bioconjugation with avidin.
Co-reporter:Daniel Wilms, Johannes Klos, Andreas F. M. Kilbinger, Holger Löwe and Holger Frey
Organic Process Research & Development 2009 Volume 13(Issue 5) pp:961-964
Publication Date(Web):June 17, 2009
DOI:10.1021/op900069a
We report on the development of an alternative protocol for the facile, solvent-free synthesis of various novel imidazolium-based ionic liquids (ILs) that affords highly pure products without the necessity of subsequent purification steps. The continuous approach is based on the combination of HPLC pumps with a micromixer and a capillary residence tube. Our system provides a high degree of control over the alkylation reactions due to a high surface-to-volume ratio and superior heat and mass transport. Within the scope of our studies, we focused on ionic liquids containing differently substituted phenyl rings and characterized these compounds with respect to further use for direct application or subsequent reaction sequences. Scale-up can conveniently be achieved by operating several reactors with high continuous throughput in parallel.
Co-reporter:Frederik Wurm;Ulrike Kemmer-Jonas
Polymer International 2009 Volume 58( Issue 9) pp:989-995
Publication Date(Web):
DOI:10.1002/pi.2619
Abstract
BACKGROUND: Until recently, hyperbranched polymers were thought to be ill-defined materials that were not useful as building blocks for well-defined complex polymer architectures. It is a current challenge to develop strategies that offer rapid access to well-defined hyperbranched block copolymers.
RESULTS: A convenient three-step protocol for the synthesis of double-hydrophilic hyperbranched–linear–hyperbranched ABA-type triblock copolymers based on poly(ethylene oxide) (PEO) and hyperbranched polyglycerol (hbPG) is presented. The Bola-type polymers exhibiting an aliphatic polyether structure were prepared from a linear (lin) linPG-b-PEO-b-linPG precursor triblock. The materials exhibit low polydispersities (Mw/Mn) in the range 1.19–1.45. The molecular weights of the block copolymers range from 6300 to 26 200 g mol−1, varying in the length of both the linear PEO chain as well as the hbPG segments. Detailed characterization of the thermal properties using differential scanning calorimetry demonstrates nanophase segregation of the blocks.
CONCLUSION: The first example of well-defined ABA hyperbranched–linear–hyperbranched triblock copolymers with PEO middle block and hbPG A-blocks is presented. The biocompatible nature of the aliphatic polyether blocks renders these materials interesting for biomedical purposes. These new materials are also intriguing with respect to their supramolecular order and biomineralization properties. Copyright © 2009 Society of Chemical Industry
Co-reporter:Florian K. Wolf and Holger Frey
Macromolecules 2009 Volume 42(Issue 24) pp:9443-9456
Publication Date(Web):November 23, 2009
DOI:10.1021/ma9016746
A series of (hyper)branched poly(l-lactide)(PLLA) copolymers has been prepared by ring-opening multibranching copolymerization of l-lactide with a hydroxyl-functional (ABB′) lactone inimer, 5HDON (5-hydroxymethyl-1,4-dioxane-2-on). Polymerization was conducted in bulk and solution and catalyzed either by stanneous-2-ethyl hexanoate (Sn(Oct)2) or an organic base, 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD). Precise structural characterization of the resulting branched copolyester structures was accomplished by a combination of 2D NMR techniques, relying on the comparison with model compounds. The 5HDON inimer was employed in 1% to 20% fractions and is incorporated either as a dendritic unit or as a focal structure, but hardly in the linear mode. A detailed reaction mechanism was derived from kinetic investigation of the polymerization via NMR spectroscopy, preparative and analytical SEC and MALDI-TOF MS. The evolution and the extent of branching have been monitored and quantified. Both the degree of branching (DB = 2D(HDON)/2D(HDON) + L(lactide); DB = 0.02−0.22) and the molecular weight (MN = 1200−34000 g/mol) could be tailored by variation of the monomer/inimer ratio. For Sn(Oct)2 catalyzed polymerization approximately 50% of the inimer is transformed into dendritic units. In the case of TBD catalysis, the formation of dendritic units was suppressed at room temperature, resulting in linear poly(lactide) functionalized with a lactone end group. The fozcal 5HDON unit of the branched structures is susceptible to further functionalization, for example, by reaction with primary hydroxyl groups, leading to branched polylactide functionalized with precisely one single dye label at the focal moiety. The formation of previously absent linear repeat units from the addition of terminal lactide units to focal 5HDON units was observed when heating the polymers above Tg for prolonged times. This reaction was accompanied by a further increase in the molecular weight of the branched copolyesters.
Co-reporter:Florian F. Wolf, Nora Friedemann and Holger Frey
Macromolecules 2009 Volume 42(Issue 15) pp:5622-5628
Publication Date(Web):July 7, 2009
DOI:10.1021/ma900894d
We describe an orthogonal polymerization strategy for the preparation of amphiphilic poly(lactide)-block-poly(2-hydroxyethyl methacrylate) (PLA-b-PHEMA) copolymers with a partially biodegradable and a potentially biocompatible polymer backbone segment. The strategy is based on an orthogonal polymerization from a double-headed initiator, which has been realized in a rapid one-pot or in a two-pot route. The “lactide first” strategy permits exclusive chain growth from the hydroxyl group of the initiator, 2-hydroxyethyl 2-bromo-2-methylpropanoate. 2-Hydroxyethyl methacrylate (HEMA) was polymerized in a second step by controlled radical polymerization (ATRP) without the use of hydroxyl protecting groups. Because of the heterogeneous character of the two blocks, ATRP had to be conducted in dimethyl sulfoxide at 80 °C, both granting sufficient solubility for the stereoregular, semicrystalline poly(lactide) block and permitting fast chain growth of the poly(HEMA) block. The PLLA/PDLA macroinitiators were synthesized using Sn(Oct)2 as a catalyst in solution (PDI = 1.07−1.17; Mn = 2000−9000 g/mol). NMR spectroscopy and MALDI-ToF MS confirmed complete terminal functionalization with the bifunctional initiator 2-hydroxyethyl 2-bromo-2-methylpropanoate. Fast growth (<10 min, 45−60% conversion) of the poly(HEMA) block was achieved with a CuCl/bipyridine or CuCl/CuCl2/bipyridene system. SEC measurements indicated complete attachment of the second block resulting in narrow polydispersity of Mw/Mn = 1.2−1.3 (Mn = 5000−9000 g/mol). Developing the concept further, removal of residual lactide monomer and Sn(Oct)2 catalyst has been proven to be redundant by a variation in the synthetic procedure. In consistence with new AGET (activators generated by electron transfer) ATRP methods, grafting of free lactide monomer onto the HEMA backbone could be avoided by oxidative deactivation of Sn(Oct)2 by small amounts of copper(II), obtaining the PLA-b-PHEMA block copolymers in one single step. DSC measurements demonstrate phase segregation of the blocks after cooling from the melt as well as work-up from solution.
Co-reporter:Frederik Wurm;Francisco Javier López Villanueva
Journal of Polymer Science Part A: Polymer Chemistry 2009 Volume 47( Issue 10) pp:2518-2529
Publication Date(Web):
DOI:10.1002/pola.23334
Abstract
A convenient two-step approach for the synthesis of ferrocenyl-functionalized long chain branched polydienes, based on both butadiene and isoprene, respectively, is presented. Classical living anionic polymerization was used to synthesize different ABn type poly(diene) macromonomers with moderate molecular weights between 1700 and 3200 g/mol and narrow polydispersity. Quantitative end-capping with chlorodimethylsilane resulted in the desired ABn macromonomer structures. In the ensuing Pt-catalyzed hydrosilylation polyaddition, branched, functionalized polydienes were obtained by a concurrent ABn + AR type of copolymerization with mono- and difunctional ferrocenyl silanes (fcSiMe2H or fc2SiMeH). Molecular weights of the branched polymers were in the range of 10,000 to 44,000 g/mol (SEC/MALLS). Because of the large number of functional end groups, high loading with ferrocene units up to 63 wt % of ferrocene was achieved. Detailed studies showed full conversion of the functional silanes and incorporation into the branched polymer. Further studies using DSC, TGA, and cyclovoltammetry (CV) measurements have been performed. Electrochemical studies demonstrated different electrochemical properties for fcSiMe2- and fc2SiMe-units. The CVs of polymers modified with diferrocenylsilane units exhibit the pattern of communicating ferrocenyl sites with two distinct, separate oxidation waves. The polymers were also deposited on an electrode surface and the electrodes investigated via CV, showing formation of electroactive films with promising results for the use of the materials in biosensors. © 2009 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem 47: 2518–2529, 2009
Co-reporter:Frederik Wurm Dipl.-Chem.;Stefan Hilf Dipl.-Chem. Dr.
Chemistry - A European Journal 2009 Volume 15( Issue 36) pp:9068-9077
Publication Date(Web):
DOI:10.1002/chem.200900666
Abstract
A convenient two-step protocol is presented for synthesis of linear-hyperbranched diblock copolymers consisting of a linear, organometallic poly(ferrocenylsilane) (PFS) block and hyperbranched poly(carbosilane) (hbPCS) segments. Linear PFS diblock copolymers were synthesized through photolytic ring-opening polymerization of dimethyl[1]silaferrocenophane as the first block and methylvinyl[1]silaferrocenophane as the second. These block copolymers served as polyfunctional cores in a subsequent hydrosilylation polyaddition of different silane-based AB2 monomers. Three AB2 monomers (methyldiallylsilane; methyldiundecenylsilane, and ferrocenyldiallylsilane) were investigated; they introduced structural diversity to the hyperbranched block and showed variable reactivity for the hydrosilylation reaction. In the case with the additional ferrocene moiety in the ferrocenyldiallylsilane monomer, an electroactive hyperbranched block was generated. No slow monomer addition was necessary for molecular-weight control of the hyperbranching polyaddition, as the core had much higher functionality and reactivity than the carbosilane monomers. Different block ratios were targeted and hybrid block copolymers with narrow polydispersity (<1.2) were obtained. All the resulting polymers were investigated and characterized by size exclusion chromatography, NMR spectroscopy, cyclic voltammetry, and TEM, and exhibited strongly anisotropic aggregation.
Co-reporter:Daniel Wilms, Frederik Wurm, Jörg Nieberle, Paul Böhm, Ulrike Kemmer-Jonas and Holger Frey
Macromolecules 2009 Volume 42(Issue 9) pp:
Publication Date(Web):April 14, 2009
DOI:10.1021/ma802701g
Hyperbranched polyglycerol (PG) is established as one of the few hyperbranched polymers that offer the possibility to control molecular weight up to Mn = 6000 g/mol. This work introduces a facile 2-step strategy that relies on the use of a low molecular weight PG (Mn = 500 and 1000 g/mol) as a macroinitiator for the slow addition of glycidol, permitting to overcome previous limitations concerning molecular weights and molecular weight control. A systematic investigation of the effect of the degree of deprotonation on the control of the polymerization reaction has been carried out. A series of hyperbranched PGs with molecular weights up to Mn = 24000 g/mol has been obtained under fully controlled conditions. The polydispersities of the samples prepared were in the range of 1.3 to 1.8. In summary, we present the first example of a synthetic strategy for a hyperbranched polymer that is now accessible over a broad range of molecular weights (300−24000 g/mol) without the ubiquitous problem of large polydispersities or the necessity for solid supports. In addition, the samples permitted a systematic study of the degree of branching DB of the hyperbranched PGs of elevated molecular weight. Values of DB = 0.60 to 0.63 were obtained, approximating the theoretical limit of 0.66 for slow monomer addition.
Co-reporter:Daniel Wilms;Johannes Klos
Macromolecular Chemistry and Physics 2008 Volume 209( Issue 4) pp:343-356
Publication Date(Web):
DOI:10.1002/macp.200700588
Co-reporter:Frederik Wurm;Francisco-J. López-Villanueva
Macromolecular Chemistry and Physics 2008 Volume 209( Issue 7) pp:675-684
Publication Date(Web):
DOI:10.1002/macp.200700574
Co-reporter:Frederik Wurm;Daniel Wilms;Johannes Klos;Holger Löwe
Macromolecular Chemistry and Physics 2008 Volume 209( Issue 11) pp:1106-1114
Publication Date(Web):
DOI:10.1002/macp.200700613
Co-reporter:Emilie Barriau;Lourdes Pastor-Pérez;Elena Berger-Nicoletti;Andreas F. M. Kilbinger;Salah-Eddine Stiriba
Journal of Polymer Science Part A: Polymer Chemistry 2008 Volume 46( Issue 6) pp:2049-2061
Publication Date(Web):
DOI:10.1002/pola.22539
Abstract
A novel series of hyperbranched polyether polyols with various n-alkyl amine cores (mono- and bifunctional) and photoactive cores (benzylamine and 1-naphthylmethylamine) have been prepared. Polymerization of glycidol was carried out in two ways, starting directly from primary amine initiators and from bisglycidolized amine initiators. NMR spectroscopy and size exclusion chromatography (SEC) showed good control over the molecular weights only, when bisglycidolized amines were used. Molecular weights and polydispersity of the hyperbranched polyglycerols prepared with these initiator-cores were in the range of 1600 to 8400 g/mol and of 1.5 to 2.5, respectively. MALDI-ToF mass spectrometry confirmed covalent attachment of the functional cores to the hyperbranched polymers. When using the bis-glycidolized amine-initiators, only functionally initiated polymers could be observed. In contrast, the direct amine-initiated polymers always showed the presence of nonfunctionalized PG homopolymer. Steady-state and time-resolved fluorescence measurements further support covalent attachment and site isolation of the functional initiators within the hyperbranched structure. © 2008 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem 46: 2049–2061, 2008
Co-reporter:Frederik Wurm, Hanna Schüle and Holger Frey
Macromolecules 2008 Volume 41(Issue 24) pp:9602-9611
Publication Date(Web):November 26, 2008
DOI:10.1021/ma8018427
The synthesis of amphiphilic linear-hyperbranched block copolymers with a linear poly(ethylene oxide) (PEO) segment and a hyperbranched poly(carbosilane)s (PCS) block in a rapid three-step strategy is described, combining oxyanionic polymerization with carbosilane chemistry. A linear precursor block copolymer was synthesized via anionic polymerization of allyl glycidyl ether onto a commercial hydroxyl-terminated PEO, using its cesium alkoxide as macro-initiator. The resulting linear AB or ABA-type di- or triblock copolymers serve as polymer cores for the subsequent hydrosilylation polyaddition of an AB2-type carbosilane monomer. Di(allyl)methylsilane or methyldi(undec-10-enylsilane) were employed, using Karstedt’s catalyst as Pt0-species. Due to the high reactivity of allyloxy groups in catalytic hydrosilylation reactions, the slow monomer addition technique, previously used for the controlled polyaddition of AB2-monomers, was not necessary in this case and complete conversion of all core allyl groups is achieved. Both the molecular weights of the hyperbranched block and the linear block were varied. The resulting polymers exhibited molecular weights up to 40,100 g/mol with rather low apparent polydispersities between 1.10 and 1.47. Phase segregation of the block copolymers was investigated by differential scanning calorimetry, demonstrating strongly segregated nanophases for all block copolymers. Transition electron microscopy showed unusual anisotropic morphologies in solution, which depended both on the length of the hyperbranched block and the nature of the carbosilane monomer used.
Co-reporter:Yi Shen;Min Kuang Dr.;Zhong Shen Dr.;Jörg Nieberle;Hongwei Duan Dr.
Angewandte Chemie International Edition 2008 Volume 47( Issue 12) pp:2227-2230
Publication Date(Web):
DOI:10.1002/anie.200704572
Co-reporter:Francisco-Javier López-Villanueva;Frederik Wurm;Andreas F. M. Kilbinger
Macromolecular Rapid Communications 2007 Volume 28(Issue 6) pp:704-709
Publication Date(Web):21 MAR 2007
DOI:10.1002/marc.200600782
A facile two-step synthesis for branched poly(isoprene)s (PI) based on polyaddition of ABn-type macromonomers is described. The synthesis of the macromonomers was achieved by anionic polymerization of isoprene and subsequent end-capping of the polymers by addition of chlorodimethylsilane to the living carbanions. This led to PI-based macromonomers with narrow polydispersity (/ < 1.15) and molecular weights in the range of 1 700 – 22 100 g · mol−1. Synthesis of the branched polymers was carried out by a hydrosilylation-based polymerization of the macromonomers. Characterization via SEC, SEC-MALLS, coupled SEC-viscosimetry and 1H-NMR-spectroscopy supported the formation of branched structures. Interestingly, these branched polymers exhibited α-values that were similar to those reported for hyperbranched polymers based on AB2-monomers.
Co-reporter:Francisco-Javier López-Villanueva;Frederik Wurm;Andreas F. M. Kilbinger
Macromolecular Rapid Communications 2007 Volume 28(Issue 6) pp:
Publication Date(Web):21 MAR 2007
DOI:10.1002/marc.200790011
Front Cover: The cover image shows the facile two-step synthesis of branched poly(isoprene)s, based on the polyaddition of ABn-type macromonomers. The macromonomers were obtained by anionic polymerization of isoprene and subsequent end-capping with chlorodimethylsilane. Subsequent hydrosilylation leads to branched polyolefins. Further details can be found in the article by F.-J. López-Villanueva, F. Wurm, A. F. M. Kilbinger, and H. Frey* on page 704.
Co-reporter:D. Wilms;J. Nieberle;J. Klos;H. Löwe;H. Frey
Chemical Engineering & Technology 2007 Volume 30(Issue 11) pp:
Publication Date(Web):26 OCT 2007
DOI:10.1002/ceat.200700277
Hyperbranched polymers have been synthesized in a microreactor for the first time, employing the known ring-opening multibranching polymerization of glycidol. Microreactors are well-known to be beneficial for highly exothermic reactions because of their capability to enhance mass and heat transfer due to short diffusion pathways and large interfacial areas per volume. The characteristics of the microstructured reaction system were utilized to engineer a continuous flow process for the preparation of well-defined hyperbranched polyglycerols with molecular weights up to 1,000 g/mol. Increased flow rates, as well as the use of highly polar solvents, led to the partial formation of very narrowly distributed (Mw/Mn = 1.05–1.15) high molecular weight fractions (Mn up to 150,000 g/mol). NMR- and MALDI-ToF spectra confirmed incorporation of the multifunctional initiator core into the hyperbranched polymer structure.
Co-reporter:Yu Chen, Holger Frey, Ralf Thomann, Salah-Eddine Stiriba
Inorganica Chimica Acta 2006 Volume 359(Issue 6) pp:1837-1844
Publication Date(Web):10 April 2006
DOI:10.1016/j.ica.2005.06.067
We report a systematic study on the encapsulation of palladium nanoparticles in optically active amphiphilic hyperbranched polyglycerols with different optical signs and different degrees of polymerization, namely (−)-P(G40C160.5) 1 and (+)-P(G73C160.5) 2. Several issues have been addressed here: (a) relatively wide size distributions (1–5 nm) of palladium nanoparticles have been achieved, (b) a remarkable template effect (1, DPn = 40, 1.2 ± 0.1 nm; 2, DPn = 73, 2.3 ± 0.1 nm average particle size) has been observed using TEM technique, as shown by the particle size dependent on the degree of polymerization of the polymers, (c) NaBH4 is found to be a convenient reducing agent to produce small particle size compared with gaseous hydrogen, (d) catalytic Heck reaction of 2,3-dihydrofuran and aryl triflate has been tested successfully without enantiocontrol.Palladium nanoparticles with relatively wide size distribution (1–5 nm) have been synthesized using optically active amphiphilic hyperbranched polyglycerols, (−)-P(G40C160.5) 1 and (+)-P(G73C160.5) 2 as polymeric templates. A remarkable template effect (1, DPn = 40, 1.2 ± 0.1 nm; 2 DPn = 73, 2.3 ± 0.1 nm average particle size) has been observed, as demonstrated by the particle size dependent on degree of polymerization of the polymers. The role of the reducing agents in the production of small particle size is presented as well as preliminary catalytic essay in a model catalytic Heck reaction.
Co-reporter:Zhong Shen;Yu Chen;Emilie Barriau
Macromolecular Chemistry and Physics 2006 Volume 207(Issue 1) pp:57-64
Publication Date(Web):19 DEC 2005
DOI:10.1002/macp.200500332
Summary: Well-defined multi-arm star block copolymers, polyglycerol-block-poly(tert-butyl acrylate) (PG-b-PtBA), with average arm-numbers of 17, 27, 36, 66 and 90 arms, respectively, have been prepared by atom transfer radical polymerization (ATRP) of tBA in acetone, using a core-first strategy. After hydrolysis with excess concentrated HCl in refluxing dioxane, full hydrolysis of the tert-butyl ester groups was achieved, resulting in multi-arm star polyelectrolytes, polyglycerol-block-poly(acrylic acid) (PG-b-PAA). The hyperbranched macroinitiators employed were prepared on the basis of hyperbranched polyglycerols via esterification with 2-bromoisobutyryl bromide. Both CuBr/PMDETA and CuBr/Me6TREN catalyst systems have been employed for ATRP of tBA. CuBr/PMDETA was found to permit good control. Polydispersity indices for the new multi-arm stars were mainly in the range of 1.22 to 1.4, and the absolute data were in agreement with the calculated values. Moreover, kinetic curves show a linear dependence of ln([M]0/[M]t) on time, confirming that the polymerizations are controlled.
Co-reporter:Alejra García Marcos;Holger Kautz;Emilie Barriau;Emilie Barriau;Alejra García Marcos;Holger Kautz
Macromolecular Rapid Communications 2005 Volume 26(Issue 11) pp:862-867
Publication Date(Web):27 MAY 2005
DOI:10.1002/marc.200500184
Summary: A convenient three-step strategy has been developed for the preparation of well-defined amphiphilic, linear-hyperbranched block copolymers by hypergrafting. The synthetic procedure is based on a combination of carbanionic polymerization with the alkoxide-based, controlled ring-opening multibranching polymerization of glycidol. A linear AB diblock copolymer polystyrene-block-polybutadiene (PS-b-PB) with narrow polydispersity was obtained by anionic copolymerization. Subsequent hydroxylation by hydroboration led to PS508-b-(PB-OH)56, used as macroinitiator for the polymerization of glycidol under slow monomer addition conditions.
Co-reporter:Holger Frey;Emilie Barriau;Holger Kautz;Alejra García Marcos;Emilie Barriau;Alejra García Marcos;Holger Kautz
Macromolecular Rapid Communications 2005 Volume 26(Issue 11) pp:
Publication Date(Web):6 JUN 2005
DOI:10.1002/marc.200590021
Co-reporter:Mario Smet;Carsten Gottschalk;Sunny Skaria
Macromolecular Chemistry and Physics 2005 Volume 206(Issue 24) pp:2421-2428
Publication Date(Web):5 DEC 2005
DOI:10.1002/macp.200500397
Summary: Hyperbranched aliphatic copolyesters have been prepared by the copolymerization of ε-caprolactone and 2,2-bis(hydroxymethyl)butyric acid (AB2-monomer), catalyzed by (i) HfCl4(THF)2 and (ii) diphenylammonium trifluoromethanesulfonate (DPAT), respectively. In both cases, copolymerization by combined ROP/AB2-polycondensation was achieved. The degree of branching (DB) and consequently the density of functional groups of the resulting copolyesters were controlled by the comonomer ratio in the feed. Molecular weights in the range = 22 000–166 000 g · mol−1 (GPC, PS standards) were obtained, with apparent polydispersity indices of 1.20 to 1.95. The DB was in the range 0.03–0.35. Remarkably, HfCl4(THF)2 appeared to cause no transesterification of the ester bonds in the hyperbranched polymer formed. Further esterification or functionalization of the hydroxyl end groups of the hyperbranched polymers is therefore possible in a convenient two step/one pot process. The prepared hyperbranched polycaprolactones can be used as multifunctional initiators for the ROP of ε-caprolactone, which is also catalyzed by HfCl4(THF)2, resulting in multi-arm star polymers. Diphenylammonium trifluoromethanesulfonate (DPAT) was also found to catalyze the combination of ROP and AB2 polycondensation. However, the applicability of this system is restricted due to side reactions that can lead to crosslinking.
Co-reporter:Christoph Tonhauser, Boris Obermeier, Christine Mangold, Holger Löwe and Holger Frey
Chemical Communications 2011 - vol. 47(Issue 31) pp:NaN8966-8966
Publication Date(Web):2011/07/07
DOI:10.1039/C1CC12956B
A series of block copolymers bearing a single amino in-chain functionality was synthesized viaanionic polymerization of styrene and ethylene oxide. By means of both a conventional and a continuous setup, living polystyrene was quantitatively end functionalized with an oxirane (DBAG) prior to the polymerization of the poly(ethylene oxide) segment. The in-chain amine was conjugated with a fluorescent dye.

![Ferrocene, [(2-oxiranylmethoxy)methyl]-](/data/chemimg/3781500/1417307-37-9.png)
![Ferrocene, [(2-oxiranylmethoxy)methyl]-](/data/chemimg/3781500/1417307-37-9_b.png)
![Propanoic acid, 3-[2-[2-(2-azidoethoxy)ethoxy]ethoxy]-](/data/chemimg/3754800/1056024-94-2.png)
![Propanoic acid, 3-[2-[2-(2-azidoethoxy)ethoxy]ethoxy]-](/data/chemimg/3754800/1056024-94-2_b.png)
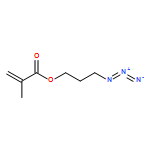
![Tricyclo[3.3.1.13,7]decane, 1-[(2-propenyloxy)methyl]-](http://img.cochemist.com/ccimg/862800/862729-17-7.png)
![Tricyclo[3.3.1.13,7]decane, 1-[(2-propenyloxy)methyl]-](http://img.cochemist.com/ccimg/862800/862729-17-7_b.png)


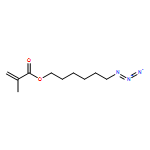
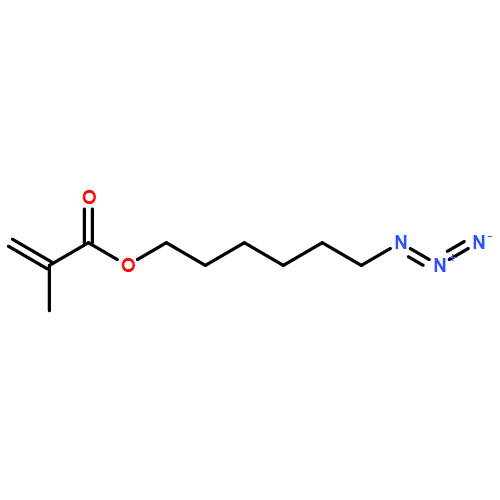


![Poly[oxy[(methoxymethyl)-1,2-ethanediyl]]](http://img.cochemist.com/ccimg/137600/137597-55-8.png)
![Poly[oxy[(methoxymethyl)-1,2-ethanediyl]]](http://img.cochemist.com/ccimg/137600/137597-55-8_b.png)
![Ethanol, 2-[2-(2-azidoethoxy)ethoxy]-](/data/chemimg/964400/86520-52-7.png)
![Ethanol, 2-[2-(2-azidoethoxy)ethoxy]-](/data/chemimg/964400/86520-52-7_b.png)
![Ethanol, 2-[2-(2-hydroxyethoxy)ethoxy]-, 1-(4-methylbenzenesulfonate)](/data/chemimg/703000/77544-68-4.png)
![Ethanol, 2-[2-(2-hydroxyethoxy)ethoxy]-, 1-(4-methylbenzenesulfonate)](/data/chemimg/703000/77544-68-4_b.png)
![OXIRANE, [(TRICYCLO[3.3.1.13,7]DEC-1-YLMETHOXY)METHYL]-](http://img.cochemist.com/ccimg/27900/27866-06-4.png)
![OXIRANE, [(TRICYCLO[3.3.1.13,7]DEC-1-YLMETHOXY)METHYL]-](http://img.cochemist.com/ccimg/27900/27866-06-4_b.png)
![Oxirane, [(2-ethoxyethoxy)methyl]-](http://img.cochemist.com/ccimg/16500/16495-58-2.png)
![Oxirane, [(2-ethoxyethoxy)methyl]-](http://img.cochemist.com/ccimg/16500/16495-58-2_b.png)
![5H-Benzo[a]phenoxazin-5-one,9-(diethylamino)-](http://img.cochemist.com/ccimg/7400/7385-67-3.png)
![5H-Benzo[a]phenoxazin-5-one,9-(diethylamino)-](http://img.cochemist.com/ccimg/7400/7385-67-3_b.png)
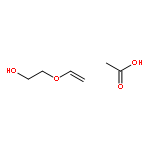
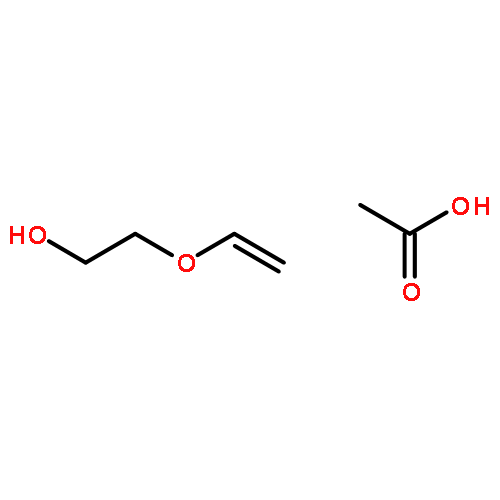
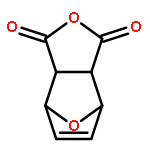
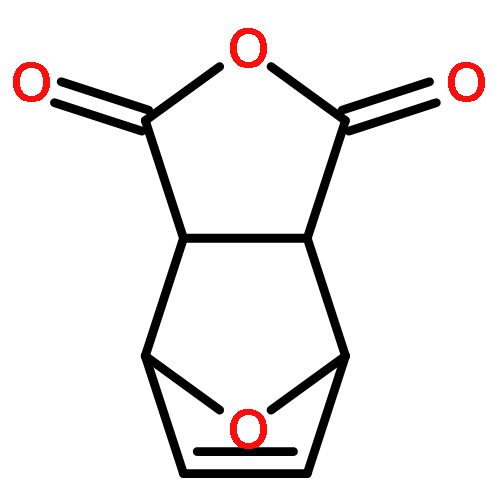
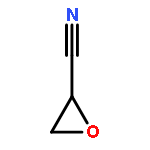
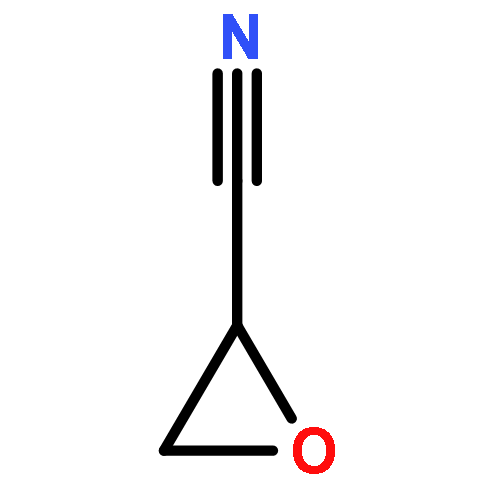
![3,5,9-Trioxa-4-phosphaheptacos-18-en-1-aminium,4-hydroxy-N,N,N-trimethyl-10-oxo-7-[[(9Z)-1-oxo-9-octadecen-1-yl]oxy]-, innersalt, 4-oxide, (7R,18Z)-](http://img.cochemist.com/ccimg/4300/4235-95-4.png)
![3,5,9-Trioxa-4-phosphaheptacos-18-en-1-aminium,4-hydroxy-N,N,N-trimethyl-10-oxo-7-[[(9Z)-1-oxo-9-octadecen-1-yl]oxy]-, innersalt, 4-oxide, (7R,18Z)-](http://img.cochemist.com/ccimg/4300/4235-95-4_b.png)
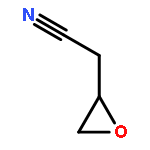
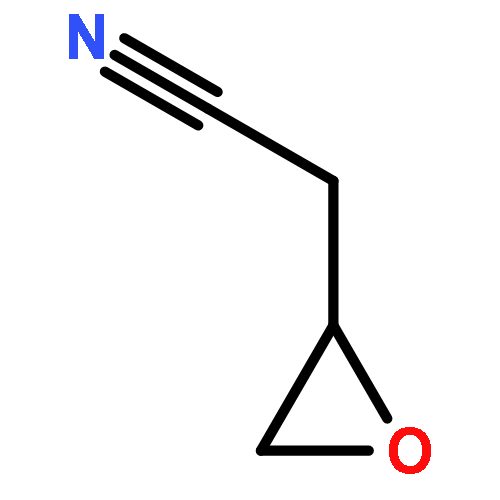

![4-[(2,3-Epoxypropoxy)methyl]-2,2-dimethyl-1,3-dioxolane](http://img.cochemist.com/ccimg/1700/1607-37-0.png)
![4-[(2,3-Epoxypropoxy)methyl]-2,2-dimethyl-1,3-dioxolane](http://img.cochemist.com/ccimg/1700/1607-37-0_b.png)