Co-reporter:Tobias Schwabe, Maarten T. P. Beerepoot, Jógvan Magnus Haugaard Olsen and Jacob Kongsted
Physical Chemistry Chemical Physics 2016 vol. 18(Issue 2) pp:1349-1349
Publication Date(Web):2015/12/15
DOI:10.1039/C5CP90225H
Correction for ‘Analysis of computational models for an accurate study of electronic excitations in GFP’ by Tobias Schwabe et al., Phys. Chem. Chem. Phys., 2015, 17, 2582–2588.
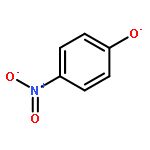
Co-reporter:Heiner Schröder, Anne Creon, and Tobias Schwabe
Journal of Chemical Theory and Computation 2015 Volume 11(Issue 7) pp:3163-3170
Publication Date(Web):May 29, 2015
DOI:10.1021/acs.jctc.5b00400
A reformulated version of Grimme’s most recent DFT dispersion correction with Becke–Johnson damping (DFT-D3(BJ)) is presented, which only depends on C6 dispersion coefficients. The role of the higher order correction terms in the DFT-D3(BJ) model is critically investigated, and a sigmoidal interpolation function for adjusting to different density functional approximations (DFA) is employed alternatively, while keeping finite damping of Becke and Johnson. For the proposed C6-only dispersion correction scheme, only one parameter needs to be fitted per DFA (instead of three for DFT-D3(BJ)). Eight standard DFAs from different classes are parametrized and evaluated. In comparison to DFT-D3(BJ), one of the most accurate corrections up to date, the new correction shows only negligible deviations in accuracy for the huge GMTKN30 benchmark set.
Co-reporter:Tobias Schwabe, Maarten T. P. Beerepoot, Jógvan Magnus Haugaard Olsen and Jacob Kongsted
Physical Chemistry Chemical Physics 2015 vol. 17(Issue 4) pp:2582-2588
Publication Date(Web):03 Dec 2014
DOI:10.1039/C4CP04524F
Using the chromophore of the green fluorescent protein (GFP), the performance of a hybrid RI-CC2/polarizable embedding (PE) model is tested against a quantum chemical cluster approach. Moreover, the effect of the rest of the protein environment is studied by systematically increasing the size of the cluster and analyzing the convergence of the excitation energies. It is found that the influence of the environment of the chromophore can accurately be described using a polarizable embedding model with only a minor error compared to a full quantum chemical description. It is also shown that the treatment of only a small region around the chromophore is only by coincidence a good approximation. Therefore, such cluster approaches should be used with care. Based on our results, we suggest that polarizable embedding models, including a large part of the environment to describe its effect on biochromophores on top of an accurate way of describing the central subsystem, are both accurate and computationally favourable in many cases.
Co-reporter:Tobias Schwabe
The Journal of Physical Chemistry B 2015 Volume 119(Issue 33) pp:10693-10700
Publication Date(Web):July 28, 2015
DOI:10.1021/acs.jpcb.5b05206
For the modeling of solvatochromism with an explicit representation of the solvent molecules, the quality of preceding molecular dynamics simulations is crucial. Therefore, the possibility to apply force fields which are derived with as little empiricism as possible seems desirable. Such an approach is tested here by exploiting the sensitive solvatochromism of p-nitroaniline, and the use of reliable excitation energies based on approximate second-order coupled cluster results within a polarizable embedding scheme. The quality of the various MD settings for four different solvents, water, methanol, ethanol, and dichloromethane, is assessed. In general, good agreement with the experiment is observed when polarizable force fields and special treatment of hydrogen bonding are applied.
Co-reporter:Tobias Schwabe
The Journal of Physical Chemistry A 2013 Volume 117(Issue 13) pp:2879-2883
Publication Date(Web):March 8, 2013
DOI:10.1021/jp401495u
In a recent paper, Xu et al. [J. Phys. Chem. A2012, 116, 11668] emphasized the importance of core–electron correlation effects to describe the Si2H6BH3 complex and related systems properly. Unexpected large energy differences between a frozen core and all electron treatment were observed. In the present study, it will be shown that these energy differences are an artifact of an insufficient choice of basis set and can be attributed to an intramolecular basis set superposition error (BSSE). Although the general problem is known, systematic studies on the effect are scarce. Therefore, the BSSE in related systems is investigated. This study shows that the problem of BSSE for core–electron correlation is quite common if inadequate basis sets are applied and that it amounts to 2 kcal mol–1 on average in binding energies for the given test set (with a maximum of 5.8 kcal mol–1).
Co-reporter:Tobias Schwabe, Kristian Sneskov, Jógvan Magnus Haugaard Olsen, Jacob Kongsted, Ove Christiansen, and Christof Hättig
Journal of Chemical Theory and Computation 2012 Volume 8(Issue 9) pp:3274-3283
Publication Date(Web):July 25, 2012
DOI:10.1021/ct3003749
We present a combination of the polarizable embedding (PE) method with the resolution-of-the-identity implementation of the approximate coupled-cluster singles and doubles method CC2. The new approach, termed PERI–CC2, allows one to study excited state phenomena of large solvated molecular systems with an accurate correlated wave function method. Central to the PE approach is the advanced description of the environmental electrostatic potential and inclusion of polarization, and the quintessence of RI-CC2 is efficient access to excited state properties while retaining the accuracy associated with CC theory. To maintain efficiency, an approximate truncated CC2 density is introduced to calculate the PE contributions. Explicitly, we derive the central equations and outline an implementation of polarizable embedding for the RI-CC2 approach. The new method is tested against previous PE–CC2 and PE–CCSD results for solvatochromic shifts, demonstrating how the important effects of polarization are incorporated well with PERI–CC2 but with a dramatically reduced overall computational cost. A follow-up investigation of the solvatochromic shift of uracil in aqueous solution further illustrates the potential of PERI–CC2. We discuss the need to explicitly incorporate several water molecules into the region treated by quantum mechanics in order to obtain a reliable and accurate description of the physical effects when specific solute/solvent interactions as, e.g., hydrogen-bonds are involved.
Co-reporter:Tobias Schwabe, Maarten T. P. Beerepoot, Jógvan Magnus Haugaard Olsen and Jacob Kongsted
Physical Chemistry Chemical Physics 2015 - vol. 17(Issue 4) pp:NaN2588-2588
Publication Date(Web):2014/12/03
DOI:10.1039/C4CP04524F
Using the chromophore of the green fluorescent protein (GFP), the performance of a hybrid RI-CC2/polarizable embedding (PE) model is tested against a quantum chemical cluster approach. Moreover, the effect of the rest of the protein environment is studied by systematically increasing the size of the cluster and analyzing the convergence of the excitation energies. It is found that the influence of the environment of the chromophore can accurately be described using a polarizable embedding model with only a minor error compared to a full quantum chemical description. It is also shown that the treatment of only a small region around the chromophore is only by coincidence a good approximation. Therefore, such cluster approaches should be used with care. Based on our results, we suggest that polarizable embedding models, including a large part of the environment to describe its effect on biochromophores on top of an accurate way of describing the central subsystem, are both accurate and computationally favourable in many cases.
Co-reporter:Tobias Schwabe, Maarten T. P. Beerepoot, Jógvan Magnus Haugaard Olsen and Jacob Kongsted
Physical Chemistry Chemical Physics 2016 - vol. 18(Issue 2) pp:NaN1349-1349
Publication Date(Web):2015/12/15
DOI:10.1039/C5CP90225H
Correction for ‘Analysis of computational models for an accurate study of electronic excitations in GFP’ by Tobias Schwabe et al., Phys. Chem. Chem. Phys., 2015, 17, 2582–2588.