Co-reporter:Illia O. Feskov, Anton V. Chernykh, Ivan S. Kondratov, Mariya Klyachina, Constantin G. Daniliuc, and Günter Haufe
The Journal of Organic Chemistry December 1, 2017 Volume 82(Issue 23) pp:12863-12863
Publication Date(Web):October 11, 2017
DOI:10.1021/acs.joc.7b02259
Hitherto unknown cis- and trans-1-amino-3-hydroxy-3-methylcyclobutanecarboxylic acids were synthesized in multigram scale. The obtained compounds can be considered as achiral conformationally restricted analogues of threonine with fixed spatial orientation of functional groups. pKa values are noticeably different for both amino acids. According to the X-ray data the cyclobutane rings in both compounds are almost planar (the corresponding torsion angles are below 7°).
Co-reporter:Katharina Bartels, Benjamin Schinor, Günter Haufe
Journal of Fluorine Chemistry 2017 Volume 203(Volume 203) pp:
Publication Date(Web):1 November 2017
DOI:10.1016/j.jfluchem.2017.09.008
•Metal catalyzed cyclopropanations of substituted α-fluorostyrenes with diazoacetate provide mixtures of cis/trans-isomeric 2-aryl-2-fluorocyclopropane carboxylates.•In situ generation of diazoacetate in aqueous solution with subsequent iron porphyrin catalyzed cyclopropanation is safe.•Cu(acac)2 catalyzed cyclopropanation α-fluorostyrene proceeds slower than with styrene.•The trans-diastereoselectivity of α-fluorostyrene is lower compared to styrene.The diastereoselectivity of cyclopropanations of styrene and α-fluorostyrene with diazoacetate depends on the catalyst used and the presence or absence of the fluorine substituent. The Cu(acac)2 catalyzed reaction of styrene with diazoacetate led to 3:1 selectivity in favor of trans-2-phenylcyclopropane carboxylate, while α-fluorostyrene gave a 1:1 mixture of cis/trans-isomers. A competition experiment proved that α-fluorostyrene reacted slower compared to styrene itself. With the bulkier tetraphenyl-iron(III)-porphyrin chloride as catalyst, 10:1 or 3:1 mixture, respectively, were obtained. An advantage of the latter protocol is the in situ formation of ethyl diazoacetate from ethyl glycinate hydrochloride in aqueous solution by diazotation avoiding the in-substance application of the potentially explosive ethyl diazoacetate. Accordingly, a series of diastereoisomeric ethyl 2-aryl-2-fluoro-cyclopropane carboxylates was synthesized from p- or m-substituted α-fluorostyrenes.Download high-res image (87KB)Download full-size image
Co-reporter:Dr. Volodymyr Kozel;Dr. Constantin-Gabriel Daniliuc; Dr. Peer Kirsch; Dr. Günter Haufe
Angewandte Chemie 2017 Volume 129(Issue 48) pp:15659-15663
Publication Date(Web):2017/11/27
DOI:10.1002/ange.201709279
AbstractEin direkter Zugang zum bisher unbekannten C3-symmetrischen Tricyclo[2.2.1.02,6]heptan-3,5,7-triol sowohl in racemischer als auch in enantiomerenreiner Form wurde ausgearbeitet. Die Umsetzung von 7-tert-Butoxynorbornadien mit Peroxycarbonsäuren führte durch transannulare π-Cyclisierung und Substitution der tert-Butoxy-Gruppe zu Gemischen der entsprechenden C1- und C3-symmetrischen 3,5,7-Triacyloxynortricyclene. Durch Sieden in Ameisensäure wurden die C1-symmetrischen Ester größtenteils in das C3-symmetrische Formiat überführt. Nachfolgende Verseifung lieferte die diastereomeren Triole, die durch fraktionierende Kristallisation getrennt wurden. Die Enantiomerentrennung des C3-symmetrischen Triols erfolgte über die diastereomeren Tri(O-methyl)mandelsäureester. Die so im Gramm-Maßstab zugänglichen enantiomerenreinen Triole wurden als Kerneinheit von Dotierstoffen für flüssigkristalline Phasen eingesetzt.
Co-reporter:Dr. Volodymyr Kozel;Dr. Constantin-Gabriel Daniliuc; Dr. Peer Kirsch; Dr. Günter Haufe
Angewandte Chemie 2017 Volume 129(Issue 48) pp:15366-15366
Publication Date(Web):2017/11/27
DOI:10.1002/ange.201710815
Das kleinste tricyclische C3-symmetrische Triol wurde in racemischer Form in einer einfachen Eintopf-Reaktion aus kommerziell leicht zugänglichen Ausgangsmaterialien und Reagenzien synthetisiert. Eine klassische Racematspaltung führte zu “Propeller-ähnlichen” Enantiomeren, die großes Anwendungspotential in der asymmetrischen Synthese, der molekularen Erkennung und den Materialwissenschaften haben. In ihrer Zuschrift auf S. 15659 liefern G. Haufe et al. mehr Details, unter anderem zur Synthese eines chiralen Dotierstoffs für Flüssigkristalle.
Co-reporter:Dr. Volodymyr Kozel;Dr. Constantin-Gabriel Daniliuc; Dr. Peer Kirsch; Dr. Günter Haufe
Angewandte Chemie International Edition 2017 Volume 56(Issue 48) pp:15168-15168
Publication Date(Web):2017/11/27
DOI:10.1002/anie.201710815
The smallest tricyclic C3-symmetric triol was obtained as a racemate in a simple one-pot synthesis from readily available commercial starting materials and reagents. Classical resolution provided the propeller-like enantiopure triols, which have a high potential for application in asymmetric synthesis, molecular recognition, and materials science. In their Communication on page 15456 ff., G. Haufe et al. provide more details, including the synthesis of a chiral dopant for liquid crystals.
Co-reporter:Dr. Volodymyr Kozel;Dr. Constantin-Gabriel Daniliuc; Dr. Peer Kirsch; Dr. Günter Haufe
Angewandte Chemie International Edition 2017 Volume 56(Issue 48) pp:15456-15460
Publication Date(Web):2017/11/27
DOI:10.1002/anie.201709279
AbstractA straightforward access to a hitherto unknown C3-symmetric tricyclic triol both in racemic and enantiopure forms has been developed. Treatment of 7-tert-butoxynorbornadiene with peroxycarboxylic acids provided mixtures of C1- and C3-symmetric 3,5,7-triacyloxynortricyclenes via transannular π-cyclization and replacement of the tert-butoxy group. By refluxing in formic acid, the C1-symmetric esters were converted to the C3-symmetric formate. Hydrolysis gave diastereoisomeric triols, which were separated by recrystallization. Enantiomer resolution via diastereoisomeric tri(O-methylmandelates) delivered the target triols on a gram scale. The pure enantiomers are useful as core units of dopants for liquid crystals.
Co-reporter:Ivan S. Kondratov;Ivan G. Logvinenko;Nataliya A. Tolmachova;Roman N. Morev;Maria A. Kliachyna;Florian Clausen;Constantin G. Daniliuc
Organic & Biomolecular Chemistry 2017 vol. 15(Issue 3) pp:672-679
Publication Date(Web):2017/01/18
DOI:10.1039/C6OB02436J
2-Amino-2-(trifluoromethoxy)butanoic acid (O-trifluoromethyl homoserine) was synthesized as a racemate and in both enantiomeric forms. The measured pKa and log D values establish the compound as a promising analogue of natural aliphatic amino acids.
Co-reporter:Anna-Lena Dreier, Bernd Beutel, Christian Mück-LichtenfeldAndrej V. Matsnev, Joseph S. ThrasherGünter Haufe
The Journal of Organic Chemistry 2017 Volume 82(Issue 3) pp:
Publication Date(Web):December 30, 2016
DOI:10.1021/acs.joc.6b02805
Earlier studies have shown that [3,3]-sigmatropic rearrangements of allyl esters are useful for the construction of fluorine-containing carboxylic acid derivatives. This paper describes the synthesis of 3-aryl-pent-4-enoic acid derivatives bearing either a pentafluorosulfanyl (SF5) or a trifluoromethyl (CF3) substituent in the 2-position by treatment of corresponding SF5- or CF3-acetates of p-substituted cinnamyl alcohols with triethylamine followed by trimethylsilyl triflate (TMSOTf). This Ireland–Claisen rearrangement delivered approximate 1:1 mixtures of syn/anti diastereoisomers due to tiny differences (<0.5 kcal/mol) both in the energy of (Z)/(E)-isomeric ester enolates and in the alternative Zimmerman–Traxler transition states of model compounds as shown by DFT calculations. Acidic reaction conditions have to be avoided since addition of the reagents in opposite sequence (first TMSOTf then Et3N) led to oligomerization of the cinnamyl SF5- and CF3-acetates. Treatment of the corresponding regioisomeric 1-phenyl-prop-2-en-1-yl acetates under the latter conditions resulted in [1,3]-sigmatropic rearrangement and subsequent oligomerization of the intermediately formed cinnamyl esters. When Et3N was added first followed by TMSOTf, no further reaction of the formed ester was detected.
Co-reporter:Florian W. Friese, Anna-Lena Dreier, Andrej V. Matsnev, Constantin G. Daniliuc, Joseph S. Thrasher, and Günter Haufe
Organic Letters 2016 Volume 18(Issue 5) pp:1012-1015
Publication Date(Web):February 24, 2016
DOI:10.1021/acs.orglett.6b00136
Aldol reactions of pentafluorosulfanyl (SF5)-substituted acetic acid esters with both aromatic and aliphatic aldehydes proceeded with excellent anti-diastereoselectivity and good to high yields using dicyclohexylchloroborane/triethylamine. This methodology enabled the synthesis of hitherto unknown α-SF5-β-hydroxy esters. Using a norephedrine-based auxiliary, high asymmetric induction was observed. The stereochemistry of products was assigned by NMR spectroscopy and proved by X-ray diffraction analysis. The intermediate enolate was identified as a highly unstable species.
Co-reporter:Dirk Ulbrich, Constantin G. Daniliuc and Günter Haufe
Organic & Biomolecular Chemistry 2016 vol. 14(Issue 9) pp:2755-2767
Publication Date(Web):29 Jan 2016
DOI:10.1039/C6OB00131A
Intending to synthesize ω,ω-difluoroalkyl amino acid derivatives by oxidative desulfurization–fluorination reactions of suitable arylthio-2-phthalimido butanoates and pentanoates, in addition to small amounts of the target products, mainly α,ω-polyfluorinated amino acid derivatives were formed by additional sulfur-assisted α-fluorination. This novel structural motif was verified spectroscopically as well as by X-ray analysis. A plausible mechanism of formation is suggested. Using a different approach, δ,δ-difluoronorvaline hydrochloride was synthesized with at least 36% enantiomeric excess via deoxofluorination of the corresponding aldehyde.
Co-reporter:Daniel C. Ramb;Andreas Lerchen;Marvin Kischkewitz;Bernd Beutel;Santos Fustero;Günter Haufe
European Journal of Organic Chemistry 2016 Volume 2016( Issue 9) pp:1751-1759
Publication Date(Web):
DOI:10.1002/ejoc.201600088
Abstract
A series of nucleophiles, including primary and secondary amines, primary alcohols, and thiols, as well as diethyl malonate and nitromethane, were added to different fluorinated Michael acceptors including 2-fluoroalk-1-en-3-ones and 2-fluoro-1-phenylprop-2-en-1-one. The resulting β-substituted α-fluoro ketones were isolated in 34–92 % yield, depending on the substrate and the nucleophile. The best yields were obtained with secondary amines and with p-methylthiophenol.
Co-reporter:Bernd Beutel, Constantin G. Daniliuc, Burkhard Riemann, Michael Schäfers, Günter Haufe
Bioorganic & Medicinal Chemistry 2016 Volume 24(Issue 4) pp:902-909
Publication Date(Web):15 February 2016
DOI:10.1016/j.bmc.2016.01.017
Fluorine-containing inhibitors of matrix metalloproteinases (MMPs) can serve as lead structures for the development of 18F-labeled radioligands. These compounds might be useful as non-invasive imaging probes to characterize pathologies associated with increased MMP activity. Results with a series of fluorinated analogs of a known biphenyl sulfonamide inhibitor have shown that fluorine can be incorporated into two different positions of the molecular scaffold without significant loss of potency in the nanomolar range. Additionally, the potential of a hitherto unknown fluorinated tertiary sulfonamide as MMP inhibitor has been demonstrated.
Co-reporter:Dirk Ulbrich, Constantin G. Daniliuc, Günter Haufe
Journal of Fluorine Chemistry 2016 Volume 188() pp:65-75
Publication Date(Web):August 2016
DOI:10.1016/j.jfluchem.2016.06.006
•Halofluorination reactions of N-protected α,β-dehydro-α-amino acid esters proceed regioselectively.•Most of the formed α-fluoro-β-halo-α-amino acid derivatives are stable until their melting point.•Reduction of the β-halogen substituent is possible.•Nucleophilic substitution reactions of the β-halogen as well as deprotection reactions failed so far.N-protected dehydroamino acid methyl esters have been converted to α-fluoro-β-halo amino acid derivatives under halofluorination reaction conditions. The influences of the nitrogen protecting group of the substrates and of the halonium electrophile on the reaction outcome and the stability of the obtained products have been studied. Furthermore, reduction of the halogen substituent (Cl or Br) under radical conditions provided a possibility for follow-up reactions. Nucleophilic substitution reactions, however, failed.Regioselective halofluorinations of aliphatic and cyclic N-protected α,β-dehydro-α-amino acid esters lead to α-fluoro-α-amino acid derivatives bearing a reactive β-halogen.
Co-reporter:Piotr Dudziński, Andrej V. Matsnev, Joseph S. Thrasher, and Günter Haufe
The Journal of Organic Chemistry 2016 Volume 81(Issue 11) pp:4454-4463
Publication Date(Web):May 9, 2016
DOI:10.1021/acs.joc.6b00550
The chemistry of the SF5CF2 moiety has been scarcely investigated. In this report, we present synthetic pathways to a variety of SF5CF2-substituted compounds starting from vinyl ethers and SF5CF2C(O)Cl. In specific chemical environments and under particular reaction conditions, the SF5CF2 moiety is unstable in downstream products resulting in the elimination of the SF5– anion and its decomposition to SF4 and F–. Surprisingly, the formed F– can attack the intermediate difluorovinyl moiety to form trifluoromethyl substituted products. This appears to happen when an intermediate neighboring group participation involving a double bond is possible. Under slightly different conditions, the reaction stops at the stage of a difluorovinyl compound.
Co-reporter:Santos Fustero, Antonio Simón-Fuentes, Pablo Barrio, and Günter Haufe
Chemical Reviews 2015 Volume 115(Issue 2) pp:871
Publication Date(Web):July 22, 2014
DOI:10.1021/cr500182a
Co-reporter:Piotr Dudziński, Andrej V. Matsnev, Joseph S. Thrasher, and Günter Haufe
Organic Letters 2015 Volume 17(Issue 5) pp:1078-1081
Publication Date(Web):February 20, 2015
DOI:10.1021/acs.orglett.5b00268
The extraordinary properties of the pentafluorosulfanyl (SF5) group attract attention of organic chemists. While numerous SF5-substituted compounds have been synthesized, the direct introduction of SF5(CF2)n moieties has remained almost unexplored. Our investigations revealed the ambiphilic character of the SF5CF2CF2 radical. Addition reactions to electron-rich or electron-deficient alkenes profit either from its electrophilic or nucleophilic properties. Thus, the readily available SF5CF2CF2Br proved to be a promising and versatile building block for the introduction of this perfluorinated moiety.
Co-reporter:Rizwan S. Shaikh; Stefanie S. Schilson; Stefan Wagner; Sven Hermann; Petra Keul; Bodo Levkau; Michael Schäfers
Journal of Medicinal Chemistry 2015 Volume 58(Issue 8) pp:3471-3484
Publication Date(Web):March 31, 2015
DOI:10.1021/jm502021d
Sphingosine-1-phosphate (S1P) is a lysophospholipid that evokes a variety of biological responses via stimulation of a set of cognate G-protein coupled receptors (GPCRs): S1P1–S1P5. S1P and its receptors (S1PRs) play important roles in the immune, cardiovascular, and central nervous systems and have also been implicated in carcinogenesis. Recently, the S1P analogue Fingolimod (FTY720) has been approved for the treatment of patients with relapsing multiple sclerosis. This work presents the synthesis of various fluorinated structural analogues of FTY720, their in vitro and in vivo biological testing, and their development and application as [18F]radiotracers for the study of S1PR biodistribution and imaging in mice using small-animal positron emission tomography (PET).
Co-reporter:Ivan S. Kondratov;Nataliya A. Tolmachova;Violetta G. Dolovanyuk;Igor I. Gerus;Constantin-Gabriel Daniliuc;Günter Haufe
European Journal of Organic Chemistry 2015 Volume 2015( Issue 11) pp:2482-2491
Publication Date(Web):
DOI:10.1002/ejoc.201500032
Abstract
Diels–Alder reactions of ethyl 3-benzamido-2-oxo-6-(trifluoromethyl)-2H-pyran-5-carboxylate (1a) with electronically different dienophiles, including cyclic enol ethers, cycloalkenes, α,β-unsaturated ketones, and terminal acetylenes, are useful for the efficient and selective three-step preparation of trifluoromethyl-containing aromatic compounds such as 3-aminobenzoic acid derivatives. We presume that the presence of the trifluoromethyl group is the main factor in determining the regioselectivity of the initial cycloaddition.
Co-reporter:Malte Behrends, Stefan Wagner, Klaus Kopka, Otmar Schober, Michael Schäfers, Sadhana Kumbhar, Mark Waller, Günter Haufe
Bioorganic & Medicinal Chemistry 2015 Volume 23(Issue 13) pp:3809-3818
Publication Date(Web):1 July 2015
DOI:10.1016/j.bmc.2015.03.078
Matrix metalloproteinases (MMPs) are involved in a number of physiological as well as pathological processes such as atherosclerosis and tumorigenesis, where an up-regulation of MMPs is predominant. Fluorinated analogues of the hydroxamate-based non-peptidic broad-spectrum MMP inhibitor (MMPI) CGS 27023A were synthesized and inhibition potencies for MMP-2 and MMP-9 in the nanomolar range were measured using fluorimetric in vitro assays. The inhibition potencies of the herein reported fluorinated MMPIs were comparable or even superior in some cases to their non-fluorinated analogues. In contrast to the lead structure, both enantiomers of fluorinated MMPs were almost equally potent. Modelling studies suggest that the core α-amino hydroxamic acid residues appear to influence the relative potencies via specific inhibitor-peptidase interactions, including short fluorine–hydrogen contacts, within the enzyme’s pockets. The binding of the essential hydroxamate group to the zinc ion is rather unaffected by the rest of the molecule. In contrast, the corresponding α-aminocarboxylic acid derivatives are 103 times less potent or were even inactive.
Co-reporter:Ivan S. Kondratov, Maksym Ya. Bugera, Nataliya A. Tolmachova, Ganna G. Posternak, Constantin G. Daniliuc, and Günter Haufe
The Journal of Organic Chemistry 2015 Volume 80(Issue 24) pp:12258-12264
Publication Date(Web):November 9, 2015
DOI:10.1021/acs.joc.5b02171
Addition reactions of perfluoroalkyl radicals to ordinary or polyfluorinated alkenes have been frequently used to synthesize perfluoroalkylated organic compounds. Here ethyl/methyl 2-bromo-2,2-difluoroacetate, diethyl (bromodifluoromethyl)phosphonate, [(bromodifluoromethyl)sulfonyl]benzene, and ethyl 2-bromo-2-fluoroacetate were involved in Na2S2O4-mediated radical additions to vinyl ethers in the presence of alcohols to give difluoro or monofluoroacetyl-substituted acetals or corresponding difluoromethylphosphonate- and (difluoromethylphenyl)sulfonyl-substituted alkyl acetals. This methodology has also been applied as a key step in the synthesis of hitherto unknown 3,3-difluoro-GABA, completing the series of isomeric difluoro GABAs. Comparison of the pKa values of 3-fluoro- and 3,3-difluoro-GABA with that of the fluorine free parent compound showed that introduction of each fluorine lead to acidification of both the amino and the carboxyl functions by approximately one unit.
Co-reporter:Stefanie S. Schilson, Petra Keul, Rizwan S. Shaikh, Michael Schäfers, Bodo Levkau, Günter Haufe
Bioorganic & Medicinal Chemistry 2015 23(5) pp: 1011-1026
Publication Date(Web):
DOI:10.1016/j.bmc.2015.01.014
Co-reporter:Panupun Limpachayaporn ; Stefan Wagner ; Klaus Kopka ; Otmar Schober ; Michael Schäfers
Journal of Medicinal Chemistry 2014 Volume 57(Issue 22) pp:9383-9395
Publication Date(Web):October 30, 2014
DOI:10.1021/jm500718e
N-Alkylated (S)-7-halogen-5-[1-(2-methoxymethylpyrrolidinyl)sulfonyl]isatins were developed as a new group of nonradioactive reference compounds for future radiotracers. Inhibitor potency studies of these compounds suggest that the binding pockets readily accommodate both the 7-halogen substituents and aliphatic side chains (methyl to n-butyl) as well as some ω-fluorinated analogues (3-fluoropropyl and 4-fluorobutyl) at the isatin nitrogen. Indeed, compared to the halogen free parent compounds, some 7-halogenated derivatives exhibited slightly improved inhibitory potencies with IC50 values up to 2.6 nM (caspase-3) and 3.3 nM (caspase-7), respectively. Moreover, the 7-position of isatin, a potential cytochrome P450 hydroxylation site, was substituted by I, Br, Cl, and F to potentially enhance the metabolic stability of isatin sulfonamides. As an example, the radiotracer [18F]39 that was produced by 19F/18F isotope exchange was shown to be stable in human blood serum after incubation at 37 °C for at least 90 min.
Co-reporter:Ivan S. Kondratov;Nataliya A. Tolmachova;Violetta G. Dolovanyuk;Igor I. Gerus;Klaus Berger;Constantin-Gabriel Daniliuc;Günter Haufe
European Journal of Organic Chemistry 2014 Volume 2014( Issue 12) pp:2443-2450
Publication Date(Web):
DOI:10.1002/ejoc.201301485
Abstract
The Diels–Alder reactions of ethyl 3-benzamido-2-oxo-6-(trifluoromethyl)-2H-pyran-5-carboxylate with ethyl vinyl ether, 2-methoxypropene, and 1-ethoxypropene were investigated. Under different reaction conditions, intermediate oxabicyclo[2.2.2]octenones, alkoxycyclohexadienes, or aromatic products [5-benzamido-2-(trifluoromethyl)benzoates] were formed. The reaction is useful for the preparation of some hitherto unknown trifluoromethyl-containing derivatives of 3-aminobenzoic acid.
Co-reporter:Michael Marhold;Christian Stillig;Rol Fröhlich ;Günter Haufe
European Journal of Organic Chemistry 2014 Volume 2014( Issue 26) pp:5777-5785
Publication Date(Web):
DOI:10.1002/ejoc.201402535
Abstract
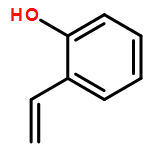
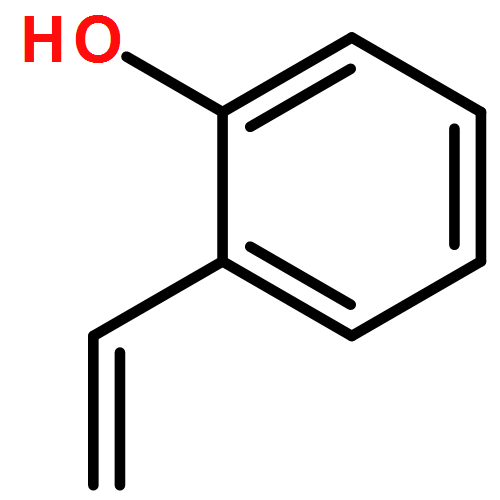
Ring-closing olefin metathesis reactions (RCM) using Grubbs II or Hoveyda's catalysts have been applied to a series of N-alkenyl-N-benzyl-α-fluoroacrylamides. α-Fluoro-α,β-unsaturated γ- or δ-lactams incorporating a fluorinated double bond were obtained in moderate to good yields, depending on the nature of substituents on the benzyl ring. The corresponding seven- and eight-membered lactams were not formed under similar conditions. When the N-benzyl group was replaced by an N-tosyl group, the corresponding ϵ-lactam was also formed in 38 % yield. When N-(2-fluoroallyl) derivatives were used instead of fluoroacryloyl derivatives, six-, seven-, and eight-membered N-heterocycles were obtained in low yields. This method was also used to synthesize fluorinated α,β-unsaturated analogues of pyrrolizidine and indolizidine alkaloids from prolinol, and also to synthesize N-benzyl-3-fluoroquinolone in three steps from commercially available 2-vinylaniline in 44 % overall yield. Also 3-fluorocoumarin and 3-fluorochromene were prepared from o-vinylphenol, and 3-fluoro-benzoxepine was available from o-allylphenol.
Co-reporter:Anna-Lena Dreier, Andrej V. Matsnev, Joseph S. Thrasher, Günter Haufe
Journal of Fluorine Chemistry 2014 Volume 167() pp:84-90
Publication Date(Web):November 2014
DOI:10.1016/j.jfluchem.2014.05.006
•New access to α-pentafluorosulfanylcarboxylic acid derivatives is disclosed.•Pentafluorosulfanylacetic acid allyl esters are used as starting materials.•[3,3]-Sigmatropic rearrangement is the key step of the synthesis.•For the first time evidence for the intermediate formation of SF5-substituted ester enolates is observed by NMR spectroscopy.A new synthetic strategy towards α-pentafluorosulfanyl carboxyl compounds was developed. Starting from allylic esters, which were prepared from fluorinated or fluorine free allylic alcohols by esterification with pentafluorosulfanylacetyl chloride, a range of trans-configured γ,δ-unsaturated α-pentafluorosulfanyl carboxylic acids were synthesized via Ireland-Claisen rearrangements. The formation of intermediate (E)- and (Z)-enolates as the key-step was confirmed by NMR spectroscopy, which also indicated that only the (Z)-enolates rearrange, while the (E)-enolates are stable under the reaction conditions and hydrolyze back to the starting allylic esters during aqueous workup. As a consequence an incomplete rearrangement of the starting esters was observed. Transformation of the formed acids into the corresponding methyl esters allowed an easier isolation of the desired pentafluorosulfanyl-containing products.[3,3]-Sigmatropic rearrangement of pentafluorosulfanylacetic acid allyl esters is used as the key step of synthesis of α-pentafluorosulfanylcarboxylic acid derivatives.
Co-reporter:Vysakh Pushpa Prasad, Stefan Wagner, Petra Keul, Sven Hermann, Bodo Levkau, Michael Schäfers, Günter Haufe
Bioorganic & Medicinal Chemistry 2014 22(19) pp: 5168-5181
Publication Date(Web):
DOI:10.1016/j.bmc.2014.08.009
Co-reporter:Panupun Limpachayaporn ; Stefan Wagner ; Klaus Kopka ; Sven Hermann ; Michael Schäfers
Journal of Medicinal Chemistry 2013 Volume 56(Issue 11) pp:4509-4520
Publication Date(Web):May 8, 2013
DOI:10.1021/jm400257a
The effector caspases-3 and -7 play a central role in programmed type I cell death (apoptosis). Molecular imaging using positron emission tomography (PET) by tracking the activity of executing caspases might allow the detection of the early onset as well as therapy monitoring of various diseases induced by dysregulated apoptosis. Herein, four new fluorinated diastereo- and enantiopure isatin sulfonamide-based potent and selective caspase-3 and -7 inhibitors were prepared by cyclic sulfate ring-opening with fluoride. All fluorohydrins exhibited excellent in vitro affinities (up to IC50 = 11.8 and 0.951 nM for caspase-3 and -7, respectively), which makes them appropriate PET radiotracer candidates. Therefore, N-(4-[18F]fluoro-3(R)-hydroxybutyl)- and N-(3(S)-[18F]fluoro-4-hydroxybutyl)-5-[1-(2(S)-(methoxymethyl)pyrrolidinyl)sulfonyl]isatin were synthesized in 140 min with 24% and 10% overall radiochemical yields and specific activities of 10–127 GBq/μmol using [18F]fluoride in the presence of Kryptofix and subsequent acidic hydrolysis. In vivo biodistribution studies in wild-type mice using PET/computed tomography imaging proved fast clearance of the tracer after tail vein injection.
Co-reporter:Panupun Limpachayaporn, Burkhard Riemann, Klaus Kopka, Otmar Schober, Michael Schäfers, Günter Haufe
European Journal of Medicinal Chemistry 2013 Volume 64() pp:562-578
Publication Date(Web):June 2013
DOI:10.1016/j.ejmech.2013.04.011
•Fluorinated inhibitors of executing caspases-3 and -7 have been synthesized.•They are isotopic reference molecules for [18F]-radiolabeled imaging agents for PET.•(S)-5-[1-(2-methoxymethylpyrrolidinyl)sulfonyl]isatin was chosen as the lead compound.•The lead was systematically substituted with fluorine in the pyrrolidine ring.•The diastereomeric 4-fluoropyrrolidinyl analogs were active in the nanomolar range.A series of new 4- or 5-substituted pyrrolidine derivatives of 5-[1-(2-methoxymethylpyrrolidinyl)sulfonyl]isatin bearing additional n-butyl or 4-fluorobutyl groups at the isatin nitrogen were prepared and their inhibitory activities have been tested against caspases-3 and -7, which are known to participate in the execution of the programmed cell death, called apoptosis. Several analogues fluorinated at the 4-position of the pyrrolidine ring were also synthesized since such inhibitors might be developed as 18F-radiotracers for molecular imaging of activated caspases in vivo by PET. Enantiomerically pure diastereomeric 4-fluoropyrrolidinyl derivatives inhibited the enzymes in the nanomolar scale, i.e.100–1000 times more efficient than the corresponding 4-methoxy analogues. The 4,4-difluorinated compound showed the best result with IC50 = 362 nM and 178 nM for the aforementioned caspases. In contrast, the 4-methoxy and 4-trifluoromethyl analogues exhibited less inhibition potencies for the enzymes in the μM scale, whereas all 4-OPEG4 (PEG4 = tetraethyleneglycol) and 5-methoxymethyl derivatives were inactive.
Co-reporter:Panupun Limpachayaporn, Michael Schäfers, Otmar Schober, Klaus Kopka, Günter Haufe
Bioorganic & Medicinal Chemistry 2013 Volume 21(Issue 7) pp:2025-2036
Publication Date(Web):1 April 2013
DOI:10.1016/j.bmc.2013.01.011
Co-reporter:Benjamin Schinor, Birgit Wibbeling, Günter Haufe
Journal of Fluorine Chemistry 2013 Volume 155() pp:102-109
Publication Date(Web):November 2013
DOI:10.1016/j.jfluchem.2013.07.003
•3- and 4-SF5-substituted benzaldehydes were transformed to the hitherto unknown styrenes by Wittig reactions.•SF5-substituted α-fluorostyrenes were synthesized by bromofluorination and subsequent HBr elimination.•Copper(I)-catalyzed reactions with ethyldiazoacetate gave diastereomeric ethyl 2-aryl-2-fluorocyclopropanecarboxylates.•The diastereomers were saponified and the acids were transformed to corresponding cyclopropylamines by Curtius degradation.•pKa values of the cyclopropanecarboxylic acids and the cyclopropylamines were determined.Starting from 3- and 4-pentafluorosulfanylbenzaldehydes, a series of cis- and trans-2-aryl-2-fluorocyclopropylamines bearing an SF5 group in 3- or 4-position of the aryl ring were synthesized in 7 steps via the corresponding cyclopropanecarboxylic acids. While the pKa values of the carboxylic acids are very little depending on the stereochemistry at the cyclopropane ring and the regiochemistry of the SF5-substituents in the aryl ring, the pKa of the corresponding 2-fluorocyclopropylamines is strongly dependent on the stereochemistry at the three-membered ring due to hyperconjugative effects of the CF and the CN bonds. Again, the regiochemistry of SF5 substitution in the phenyl ring has almost no influence.3- and 4-SF5-substituted 2-fluoro-2-phenylcyclopropylamines were synthesized from the corresponding benzaldehydes in 7 steps and their pKa values were determined.
Co-reporter:Martin Lübke, Manfred Jung, Günter Haufe
Journal of Fluorine Chemistry 2013 Volume 152() pp:144-156
Publication Date(Web):August 2013
DOI:10.1016/j.jfluchem.2013.03.011
•Artificial amino acids bearing a fluorovinyl moiety were synthesized.•Alkylation of glycine ester imines was used as key step.•γ- and δ-Fluoro-α-amino acids were used as building blocks.•Fluorinated analogs were tested as histone deacetylase inhibitors.•A terminal fluorovinyl moiety is anticipated as a bioisostere of a terminal amide group.A series of twelve fluorinated and non-fluorinated potential histone deacetylase inhibitors 25 was synthesized and their inhibitory activity was tested against rat liver histone deacetylase. The new inhibitors involve an enzyme binding element consisting of asparagine, glutamine or different short chain fluorinated or unfluorinated amino acids, a suberoyl spacer and a hydroxamic acid functionality, which is responsible for the inhibitory activity. The 4-fluoro-2-aminobutyric acid esters 1a,b, their 2-methyl derivatives 2a,b and the 2-amino-4-fluoropent-4-enoic acid esters 3a,b were synthesized by alkylation of glycine or alanine ester imides with bromofluoroethane or 2-fluoroallylbromide, respectively. Methyl 2-amino-5-fluorohex-5-enoate (4a) was prepared using 3-fluorobut-3-enyl tosylate or the iodide as alkylating reagents. An alternative pathway starting from Boc protected 3-iodo-l-alanine was more efficient. The latter method was also applied to synthesize the parent unfluorinated compound 4c using allylbromide as the alkylating reagent. The fluorinated compounds, tested as histone deacetylase inhibitors were slightly less active than comparable (S)-valine, (S)-phenylalanine or (S)-allylglycine derivatives that do not contain a fluorine atom. Interestingly, it was shown for the first time that the fluoro vinyl group which was proposed to be an aprotic amide mimic due to its electronic properties may serve as a bioisosteric replacement of a primary amide function. For the fluorinated analogs 25f (IC50 0.88 μM) and 25i (IC50 1.02 μM) a similar or an even enhanced inhibitory activity was observed compared to the unfluorinated parents 25g (IC50 0.67 μM) and 25j (IC50 1.79 μM) or the (S)-asparagine or (S)-glutamine derivatives 25l (IC50 2.10 μM) and 25m (IC50 3.90 μM).The selective synthesis of γ- and δ-fluoro-α-amino acids as well as their application as building blocks for the construction of new histone deacetylase inhibitors is reported.
Co-reporter:Peter Persich, Julia Kerschbaumer, Sandra Helling, Barbara Hildmann, Birgit Wibbeling, and Günter Haufe
Organic Letters 2012 Volume 14(Issue 22) pp:5628-5631
Publication Date(Web):November 8, 2012
DOI:10.1021/ol302820c
Transannular O-heterocyclization is applied as a key step in a total synthesis. This highly stereoselective and metal-free transformation introduces four stereocenters in one step. It was chosen to be the pivotal step in the synthesis of Murisolin and 16,19-cis-Murisolin, two annonaceous acetogenins. The efficiency of this synthesis is further illustrated by a stereodivergent late-stage separation of both synthetic routes.
Co-reporter:Ivan S. Kondratov, Violetta G. Dolovanyuk, Nataliya A. Tolmachova, Igor I. Gerus, Klaus Bergander, Roland Fröhlich and Günter Haufe
Organic & Biomolecular Chemistry 2012 vol. 10(Issue 44) pp:8778-8785
Publication Date(Web):04 Sep 2012
DOI:10.1039/C2OB26176F
The hitherto unreported reactions of β-alkoxyvinyl polyfluoroalkyl ketones with ethyl isocyanoacetate and equimolar amounts of potassium-tert-butoxide proceeded mainly in the β-position of the α,β-unsaturated ketones in cases of α-nonsubstituted 1a–e and α-methyl substituted ketones 1g–j. Other α- or β-substituted ketones 1f,k–o gave mainly products 4 of initial attack at the carbonyl carbon. Depending on the solvent, the major products of β-attack do exist in different tautomeric forms. Generally the open-chain enol tautomers 5 predominate in the polar DMSO-d6, while the cyclic γ-hemiaminals 8 are the major tautomers in the less polar CDCl3. Acid treatment of the latter compounds 8 led to the hitherto unknown ethyl 5-polyfluoroalkyl-pyrrole-2-carboxylates 11 by elimination of formic acid. Catalytic hydrogenation of pyrrole 11a was used for the synthesis of earlier unknown 5-trifluoromethyl proline 16.
Co-reporter:Ionel Humelnicu, Ernst-Ulrich Würthwein and Günter Haufe
Organic & Biomolecular Chemistry 2012 vol. 10(Issue 10) pp:2084-2093
Publication Date(Web):07 Dec 2011
DOI:10.1039/C2OB06492H
Quantum chemical calculations (DFT, SCS-MP2) show that the relative energies of the four principal alanine conformations are only marginally altered by the introduction of a single fluorine substituent into the methyl group. The fluorinegauche effect and attractive interactions of fluorine to the O–H or N–H moieties (formation of hydrogen bridges) do stabilize particular conformers of 3-fluoroalanine. This is true for the neutral molecule both in the gas phase and in aqueous solution (CPCM-model), but also for the zwitterionic forms and the conformers of the related carboxylate ions and also for the respective ammonium ions in aqueous solution. In water (CPCM calculations), the zwitterion is almost equal in energy to the most stable conformer of the neutral 3-fluoroalanine. Compared to alanine the atomic charges of the amino group and the carboxyl function of 3-fluoroalanine are not significantly influenced by the fluorine at C3, which relates to the fact that both experimental pKa values are almost equal for alanine and 3-fluoroalanine.
Co-reporter:Verena Hugenberg, Günter Haufe
Journal of Fluorine Chemistry 2012 Volume 143() pp:238-262
Publication Date(Web):November 2012
DOI:10.1016/j.jfluchem.2012.06.015
Fluorine containing compounds hold huge promise for pharmaceuticals and agrochemicals due to their specific therapeutic potency or pesticide properties. Therefore, the development of selective and efficient methods for the introduction of fluorine or fluorinated groups into organic molecules is one of the most important tasks in organofluorine chemistry today.Oxidative fluorinations of sulfur compounds like the fluoro-Pummerer rearrangement and analogous transformations such as oxidative desulfurization–fluorination reactions reveal mild, selective and efficient pathways toward mono-, di- or trifluorinated organic compounds. This article summarizes the synthetic approaches as well as the scope and limitations of fluoro-Pummerer rearrangements, oxidative desulfurization–fluorination, as well as the oxidative desulfurization–di- and trifluorination reactions. Application of these oxidative fluorination methods gives rise to various α-fluorinated sulfides, gem-difluorides and trifluoromethylated compounds.Graphical abstractMethods for the preparation of fluorine compounds by fluoro-Pummerer rearrangement of thioethers and related transformations such as various desulfurization–fluorination reactions are reviewed.Highlights► Fluoro-Pummerer rearrangement and related reactions are reviewed. ► α-Fluorothioethers are available from sulfoxides by reaction with an electrophile in the presence of a fluoride donor. ► Desulfurization–difluorination is highlighted as a method for the preparation of gem-difluorides. ► Synthesis of gem-difluorides and trifluoromethyl derivatives by oxidative desulfurization–fluorination.
Co-reporter:Dr. Günter Haufe;Satoru Suzuki;Hiroyuki Yasui;Chisato Terada;Takashi Kitayama;Dr. Motoo Shiro;Dr. Norio Shibata
Angewandte Chemie International Edition 2012 Volume 51( Issue 49) pp:12275-12279
Publication Date(Web):
DOI:10.1002/anie.201207304
Co-reporter:Dr. Günter Haufe;Satoru Suzuki;Hiroyuki Yasui;Chisato Terada;Takashi Kitayama;Dr. Motoo Shiro;Dr. Norio Shibata
Angewandte Chemie 2012 Volume 124( Issue 49) pp:12441-12445
Publication Date(Web):
DOI:10.1002/ange.201207304
Co-reporter:Wibke S. Husstedt;Susanne Wiehle;Christian Stillig;Klaus Berger;Stefan Grimme ;Günter Haufe
European Journal of Organic Chemistry 2011 Volume 2011( Issue 2) pp:355-363
Publication Date(Web):
DOI:10.1002/ejoc.201001224
Abstract
Selective syntheses of enantiopure regio- and diastereomeric methyl 2,3-fluoro-hydroxyalkanoates via four different routes employing two types of fluorination reagents are reported. The anti- and syn-3-fluoro-2-hydroxyalkanoates 1 and 3 were prepared by treating the corresponding epoxides with Olah's reagent (Py·9HF). Cyclic sulfates prepared from the enantiomeric diols were ring-opened with TBAF to give the anti- and syn-2-fluoro-3-hydroxyalkanoates 2 and 4. Thestereochemical analysis was performed mainly by NMR spectroscopy. Applying DFT/B3LYP and SCS-MP2 quantum chemical methods, the coupling constants and relative energies of conformers were calculated. Solvent effects were considered using the COSMO continuum model.
Co-reporter:Vasiliy M. Muzalevskiy, Aleksey V. Shastin, Elizabeth S. Balenkova, Günter Haufe, Valentine G. Nenajdenko
Journal of Fluorine Chemistry 2011 Volume 132(Issue 12) pp:1247-1253
Publication Date(Web):December 2011
DOI:10.1016/j.jfluchem.2011.06.030
A novel synthetic approach towards α-trifluoromethyl-phenethylamines was elaborated by reduction of the electron deficient β-aryl-α-trifluoromethyl enamines and imines with sodium cyanoborohydride in the presence of trifluoroacetic acid. The starting imines were prepared by the reaction of primary amines with β-aryl-α-trifluoromethyl enamines or β-chloro-β-(trifluoromethyl)styrenes.Graphical abstractA novel synthetic approach towards α-trifluoromethyl-phenethylamines was elaborated by reduction of the electron deficient β-aryl-α-trifluoromethylenamines and imines with sodium cyanoborohydride in the presence of trifluoroacetic acid.Highlights► New synthetic pathways towards α-trifluoromethylated phenylethylamines are disclosed. ► Corresponding enamines and β-chloro-β-trifluoromethylstyrenes are used as starting materials. ► Two pharmacophores, the β-phenethylamine and the CF3-groups, are present in the products. ► Saturated trifluoromethylated alcohols are produced as side products. ► Possible mechanisms of formation of side products are discussed.
Co-reporter:Verena Hugenberg, Roland Fröhlich and Günter Haufe
Organic & Biomolecular Chemistry 2010 vol. 8(Issue 24) pp:5682-5691
Publication Date(Web):21 Oct 2010
DOI:10.1039/C0OB00560F
An oxidative desulfurization–fluorination protocol has been used to synthesize (2S)-2-(difluoromethyl)-N-tosylpyrrolidine (6a) and (2S)-2-(trifluoromethyl)-N-tosylpyrrolidine (7a) from the (2S)-prolinol-derived (2S)-2-(4-chlorophenylthiomethyl)-N-tosylpyrrolidine (9) or (2S)-2-(dithian-2-yl)-N-tosylpyrrolidine (5). Efforts to prepare 3,3-difluoroalanine similarly from an N-protected S-aryl-cysteine ester 17 gave only traces of the target compound 18. Instead, an unique N-(α,α-difluorobenzyl)-N-α′,α′-dibromoglycine ester 19 was formed by an unprecedented sequence of reaction steps. A plausible mechanism is suggested involving a sulfur-assisted deoxygenation-difluorination of an imino oxygen and a haloform reaction like carbon–carbon bond fission as key-steps. Efforts to prepare (2S)-2-(fluoromethyl)-N-tosylpyrrolidine (12) from (2S)-N-tosylprolinol (3) by treatment with Fluolead™ (1-tert-butyl-4-trifluorosulfanyl-3,5-dimethylbenzene) gave only 5% of the target compound, but 95% of (3R)-3-fluoro-N-tosylpiperidine (11a) by ring enlargement.
Co-reporter:Verena Hugenberg, Günter Haufe
Journal of Fluorine Chemistry 2010 Volume 131(Issue 9) pp:942-950
Publication Date(Web):September 2010
DOI:10.1016/j.jfluchem.2010.06.010
Alkyl 2-arylthio-2,2-difluoroacetates are synthesized in 52–77% yield from alkyl 2-(arylthio)acetates via two succeeding fluoro-Pummerer rearrangements using the reagents combination of N-haloimides as electrophiles and excess Py·9HF as the fluoride source at room temperature. Subsequent treatment of the formed fluorinated thioethers with the same reagents at elevated temperature gave alkyl trifluoroacetates in almost quantitative yield under optimised conditions by oxidative desulfurization–fluorination.Alkyl 2-arylthio-2,2-difluoroacetates were synthesized from alkyl 2-(arylthio)acetates using the reagents combination of N-haloimides and excess Py·9HF at room temperature. Subsequent treatment of the formed fluorinated thioethers with the same reagents at elevated temperature gave alkyl trifluoroacetates by oxidative desulfurization–fluorination.
Co-reporter:Nirupam Purkayastha, Deepak M. Shendage, Roland Fröhlich and Günter Haufe
The Journal of Organic Chemistry 2010 Volume 75(Issue 1) pp:222-225
Publication Date(Web):December 7, 2009
DOI:10.1021/jo901872a
An unprecedented cascade of reactions after acid-catalyzed hydrolysis of tert-butyl (2S,5S)-2-tert-butyl-5-(2-fluoroallyl)-3-methyl-4-oxoimidazolidine-1-carboxylate 3a leading to pipecolic acid derivative 5 is presented. The vinylfluoro group is shown to be an acetonyl cation equivalent under acidic conditions. Interestingly, vinylchloro and vinylbromo groups do not show such transformation under the same conditions. The pipecolic acid derivative 5 produced in this way is further used to synthesize (2R,4R,6S)-6-tert-butyl-4-hydroxypiperidine-2-carboxylic acid 9.
Co-reporter:Anil K. Podichetty ; Stefan Wagner ; Sandra Schröer ; Andreas Faust ; Michael Schäfers ; Otmar Schober ; Klaus Kopka
Journal of Medicinal Chemistry 2009 Volume 52(Issue 11) pp:3484-3495
Publication Date(Web):May 15, 2009
DOI:10.1021/jm8015014
Caspases are responsible for the execution of the cell death program and are potentially suitable targets for the specific imaging of apoptosis in vivo. A series of N-1-substituted analogues of the small molecule nonpeptide caspase inhibitor (S)-5-[1-(2-methoxymethylpyrrolidinyl)sulfonyl]isatin (1), which may be useful for the development of caspase-targeted radioligands, were synthesized and their inhibition potencies were evaluated in vitro. Two of the most powerful techniques to introduce fluorine into organic compounds, viz, bromofluorination of olefins and fluorohydrin synthesis by ring-opening of epoxides, were used. Most of the target compounds are potent inhibitors of the two effector caspases-3 and -7. Furthermore, the 18F-radiolabeled model compound (S)-1-[4-(1-[18F]fluoro-2-hydroxyethyl)benzyl]-5-[1-(2-methoxymethyl-pyrrolidinyl)sulfonyl]isatin ([18F]37), a putative tracer for the noninvasive imaging of apoptosis by positron emission tomography (PET) was synthesized by nucleophilic epoxide ring-opening of its precursor 36. The radiochemistry utilized in the 18F-fluorination reverted to carrier-added [18F]Et3N·3HF, a new fluorine-18 source for radiolabeling.
Co-reporter:Anil Kumar Podichetty, Andreas Faust, Klaus Kopka, Stefan Wagner, Otmar Schober, Michael Schäfers, Günter Haufe
Bioorganic & Medicinal Chemistry 2009 Volume 17(Issue 7) pp:2680-2688
Publication Date(Web):1 April 2009
DOI:10.1016/j.bmc.2009.02.048
A series of new N-substituted (S)-5-[1-(2-methoxymethylpyrrolidinyl)sulfonyl]isatin derivatives has been synthesized and tested as inhibitors of caspases-3 and -7, which are known to be downstream enzymes critical in the execution of apoptosis. N-Propyl- and N-butyl isatins, as well as the corresponding terminal alcohols and regioisomeric fluorobutyl derivatives were shown to be excellent inhibitors having different binding potencies for caspases-3 and -7. In contrast, the corresponding fluoroethyl and fluoropropyl compounds were about 100–1000 times less active. Fluorinated N-benzyl isatins as well as trifluoroalkyl and difluoroalkyl derivatives were moderate inhibitors. However, isatins bearing different alkylether groups at N-1 are very weak or not active as inhibitors of caspases-3 and -7.A series of new (S)-5-[1-(2-methoxymethylpyrrolinyl)sulfonyl]isatins bearing fluorinated and non-fluorinated N-alkyl- and N-benzyl substituents has been synthesized and their inhibition potencies were assayed for recombinant human caspases-3 and -7. Among the most active inhibitors in this series are the n-propyl- and n-butyl derivatives as well as the corresponding terminal alcohols and fluorides.
Co-reporter:Nataliya A. Tolmacheva;Igor I. Gerus;Violetta G. Dolovanyuk;Ivan S. Kondratov;Günter Haufe
European Journal of Organic Chemistry 2009 Volume 2009( Issue 29) pp:5012-5019
Publication Date(Web):
DOI:10.1002/ejoc.200900684
Abstract
New δ-(polyfluoroalkyl)-δ-hydroxy-α-amino acids were synthesized from the corresponding starting 3-(benzoylamino)-6-(polyfluoroalkyl)-2H-pyran-2-ones. The key step of the synthesis was the hydrogenation of the pyrone ring. Stereoselectivity and yields depended dramatically on the reaction conditions and the nature of the polyfluoroalkyl group. Various conditions were used for the preparation of both mixtures of diastereomers and pure diastereomers of the target amino acids. The obtained δ-(polyfluoroalkyl)-δ-hydroxy-α-amino acids are of interest as analogues of 2-amino-5-hydroxyvaleric acid and glutamic acid.(© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2009)
Co-reporter:Isabella Hyla-Kryspin, Stefan Grimme, Svenja Hruschka and Günter Haufe
Organic & Biomolecular Chemistry 2008 vol. 6(Issue 22) pp:4167-4175
Publication Date(Web):22 Sep 2008
DOI:10.1039/B810108F
Fluorine substituents in organic molecules do dramatically influence the electronic structure of neighbouring functional groups and the conformation of molecules. Hence the presence of fluorine in a compound changes its chemical reactivity and biological activity. On the basis of MP2 and SCS-MP2 calculations, we discuss the conformational preferences and basicity of the diastereoisomeric 2-fluorocyclopropylamines (cis-2 and trans-2) in comparison to those of cyclopropylamine (1) and 2-fluoroethylamine (3). 1 and 2 are viewed as model compounds for the antidepressant drug tranylcypromine (trans-2-phenylcyclopropylamine, 1′a) and its fluorinated derivatives 2′. The potential energy profile for the rotation of the amino group in cis-2 differs from that of trans-2 and 1 which have very similar rotational curves. For 2 the global minimum conformer is trans-2a and the lowest energy cis-conformer 2c is less stable by 2.57 kcal mol−1. The calculated enthalpy differences between the conformers gauche-1b and s-trans-1a (2.0 kcal mol−1) as well as between gauche-3b and gauche-4a (0.2 kcal mol−1) agree well with the available experimental data of 2.0 kcal mol−1 and 0.1 ± 0.3 kcal mol−1, respectively. The calculated gas phase proton affinities (PA) of 1 (217.6 kcal mol−1), cis-2c (215.6 kcal mol−1), and trans-2a (209.3 kcal mol−1) follow the trends of the pKa values measured in solution for the diastereomeric 2-phenylcyclopropylamines 1′a and 1′b and their fluorinated derivatives cis-2′ and trans-2′. It is shown that the conformational preferences and basicity of the investigated molecules are due to stereoelectronic effects from hyperconjugative interactions which lead to different local charge distributions and different hybridization of the nitrogen lone-pair. The basicity of gauche-3a (PA = 215.3 kcal mol−1) and anti-3b (PA = 210.1 kcal mol−1) is controlled by the charge of the nitrogen atom, while that of cis-2c and trans-2a is overlap controlled as a result of different hybridization of the nitrogen lone-pair [sp4.34 (cis-2c), sp4.07 (trans-2a)].
Co-reporter:Svenja Hruschka, Thomas C. Rosen, Shinichi Yoshida, Kenneth L. Kirk, Roland Fröhlich, Birgit Wibbeling, Günter Haufe
Bioorganic & Medicinal Chemistry 2008 Volume 16(Issue 15) pp:7148-7166
Publication Date(Web):1 August 2008
DOI:10.1016/j.bmc.2008.06.048
A series of racemic, diastereoisomeric aryl cyclopropylamines substituted with fluorine in the 2-position and electron-donating and electron-withdrawing groups on the aromatic ring have been prepared. These represent analogues of the classic MAO inhibitor tranylcypromine (trans-2-phenylcyclopropylamine, 1). Their activities as inhibitors of recombinant human liver monoamine oxidases A (MAO A) and B (MAO B) were determined. The trans-compounds were low micromolar inhibitors of both MAO A and MAO B with moderate MAO A selectivity while the less active cis-analogues were MAO B selective. In the trans-series, electron-withdrawing para-substituents increased the potency of MAO A inhibition while electron-donating groups such as methyl or methoxy had no influence on this activity. In contrast, aromatic ring substitution in the trans-series had essentially no effect on the inhibition of MAO B. The corresponding cis-compounds were shown to be 10–100 times less active against MAO A, while trans- and cis-compounds were quite similar in terms of inhibition of MAO B. The best MAO A/MAO B selectivity (7:1) in the trans-series was found for trans-2-fluoro-2-(para-trifluoromethylphenyl)cyclopropylamine (7d), while a 1:27 selectivity was found for cis-2-fluoro-2-(para-fluorophenyl)cyclopropylamine (10c). These results are discussed in connection with the pKa and log D values, the mechanism of action of tranylcypromines, and the geometry of the active site of the enzymes.Electron-withdrawing para-substituents increase the potency of MAO A inhibition in the trans-series, while electron-donating groups have a weak influence on this activity. In contrast, aromatic ring substitution has essentially no effect on inhibition of MAO B. The corresponding cis-compounds are 10–100 times less active.
Co-reporter:Vasiliy M. Muzalevskiy, Valentine G. Nenajdenko, Aleksey V. Shastin, Elizabeth S. Balenkova, Günter Haufe
Journal of Fluorine Chemistry 2008 Volume 129(Issue 10) pp:1052-1055
Publication Date(Web):October 2008
DOI:10.1016/j.jfluchem.2008.06.014
A novel pathway towards trifluoromethylalcohols by an unexpected reaction of tert-butoxy-β-(trifluoromethyl)styrenes or corresponding trifluoromethylbenzyl ketones under the conditions of the Leuckart–Wallach reaction was elaborated.
Co-reporter:Svenja Hruschka, Shinichi Yoshida, Kenneth L. Kirk, Günter Haufe
Journal of Fluorine Chemistry 2008 Volume 129(Issue 9) pp:875-880
Publication Date(Web):September 2008
DOI:10.1016/j.jfluchem.2008.06.023
Diastereomeric arylcyclopropylamines substituted with fluorine in the 2-position and with electron donating or electron withdrawing groups at the aromatic ring were evaluated as inhibitors of microbial tyramine oxidase. The trans-isomers were consistently more potent inhibitors of the enzyme than the cis-isomers. Electron donating substituents increased the potency of tyramine oxidase inhibition, while electron withdrawing substituents decreased the activity. The results obtained are discussed in terms of pKa and log D values of the inhibitors as well as the mechanism of action of tranylcypromines and the geometry of the active site of the enzyme.Diastereomeric 2-aryl-2-fluorocyclopropylamines with electron-donating or electron withdrawing groups such as methox- or trifluoromethyl groups in the aromatic ring are investigated as tyramine oxidase inhibitors.
Co-reporter:Gergana S. Nikolova, Li Zhang, Xiaodong Chen, Lifeng Chi, Günter Haufe
Colloids and Surfaces A: Physicochemical and Engineering Aspects 2008 Volume 317(1–3) pp:414-420
Publication Date(Web):20 March 2008
DOI:10.1016/j.colsurfa.2007.11.013
Different methods for the selective synthesis of diastereomeric ethyl 2-fluoroalk-2-enoates 1 and 2 and 2-fluoroalk-2-en-1-ols 3 and 4, derived from the esters by reduction, with 16 and 18 carbon atoms were developed. The phase behavior of the synthesized compounds at the air/water interface was investigated by Langmuir film balance measurements and Brewster angle microscopy (BAM) and compared to that of the corresponding non-fluorinated parent compounds. While the phase behavior of the esters is dominated by the influence of the ethyl moiety in the head group and the configuration of the double bond, in the allylic alcohols the presence or absence of a vinylic fluorine substituent is crucial for the morphology and the stability of the monolayers. In addition, in the case of alcohols the fluorine prevents the crystallization.
Co-reporter:Jan Hajduch;Oldřich Paleta;Jaroslav Kvíčala
European Journal of Organic Chemistry 2007 Volume 2007(Issue 30) pp:5101-5111
Publication Date(Web):7 AUG 2007
DOI:10.1002/ejoc.200700543
3-Fluorofuran-2(5H)-one (1) and three 3,4-difluorofuran-2(5H)-ones 2–4, α,β-unsaturated lactones possessing fluorinated double bonds, were applied as dienophiles in Diels–Alder reactions with normal electron demand using diphenylisobenzofuran or cyclopentadiene as dienes. In the same reactions, furan or 2,3-dimethylbuta-1,3-diene were completely unreactive. Three structural factors of furan-2(5H)-ones appeared to have an effect on the reactivity, regioselectivity and diastereoselectivity of the [4+2] cycloadditions: the number of fluorine atoms attached to the double bond and the number and the bulkiness of alkyl substituents at the 5-position of the furan-2(5H)-one system. The monofluorinated furan-2(5H)-one 1 was generally more reactive than the difluorinated furan-2(5H)-ones 2–4. While the reactions of the furan-2(5H)-ones 2–4 with isobenzofuran exclusively gave exo products, those of the monofluorinated lactone 1 led to mixtures of endo and exo diastereoisomeric [4+2] cycloadducts. All fluorinated furan-2(5H)-ones 1–4 formed mixtures of diastereoisomeric 1:1 and 1:2 adducts with cyclopentadiene. DFT calculations of the transition states of the above Diels–Alder reactions using the BMK functional, a third-generation hybrid functional tailored for transition state calculations, together with the polarization consistent aug-pc-2 basis set, confirmed the preferential formation of the exo adducts from difluorinated furan-2(5H)-ones, while for nonfluorinated analogues a small but significant preference for the endo adducts was confirmed.(© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2007)
Co-reporter:Valentine G. Nenajdenko, Vasiliy M. Muzalevskiy, Aleksey V. Shastin, Elizabeth S. Balenkova, Günter Haufe
Journal of Fluorine Chemistry 2007 Volume 128(Issue 7) pp:818-826
Publication Date(Web):July 2007
DOI:10.1016/j.jfluchem.2007.02.014
A novel synthetic method for the preparation of α-fluoro- and the still unknown α-trifluoromethylacrylonitriles is elaborated. The reaction of α-fluorovinylbromides and α-trifluoromethylvinylbromides with CuCN leads to the title compounds in good to high yields. While the α-fluoroacrylonitriles were isolated as mixture of Z/E-isomers, the α-trifluoromethylacrylonitriles were obtained as pure Z-isomers. The α-trifluoromethylacrylonitriles are shown to be excellent dienophiles for Diels–Alder reactions.
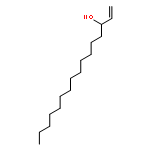
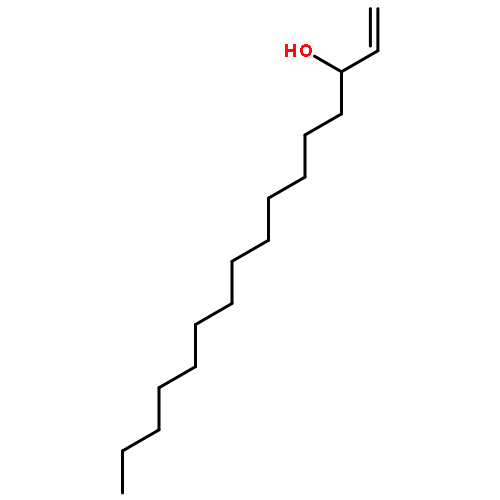
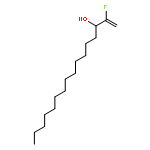
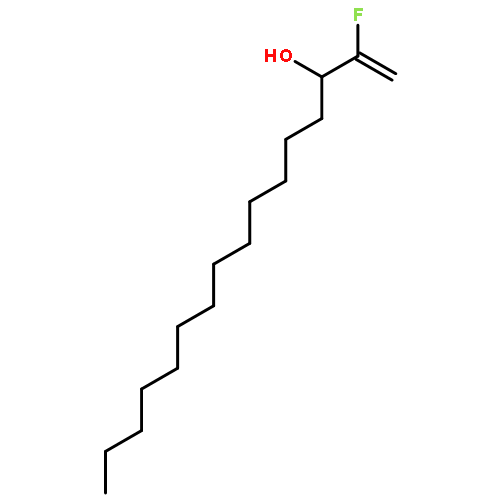
Co-reporter:Michael Marhold, Ulrich Wittmann, Stefan Grimme, Tamiko Takahashi, Günter Haufe
Journal of Fluorine Chemistry 2007 Volume 128(Issue 10) pp:1306-1317
Publication Date(Web):October 2007
DOI:10.1016/j.jfluchem.2007.05.017
A series of enantioenriched long chain 2-fluoroalk-1-en-3-ols 1 has been prepared by lipase-catalyzed resolution of the racemic compounds synthesized from terminal alkenes. The lipase of Candida antarctica was shown to be the most efficient one in terms of enantioselectivity. Transesterification of the fluorinated allylic alcohols 1 was superior over the hydrolysis in a phosphate buffer of the corresponding acetates 2. Lipase-catalyzed acetylation of allylic alcohols 1 in organic medium gave (S)-(−)-3-acetoxy-2-fluoroalk-1-enes of chain lengths C10, C16 and C18 with 68–89% yield and 92–96% ee, while the remaining (R)-(+)-2-fluoroalk-1-en-3-ols were isolated with 54–96% yield and 72–86% ee. The absolute configuration was assigned by comparison of measured and calculated CD-spectra, and unambiguously by 1H and 19F NMR spectroscopy using a modified Mosher's method. From the optically active fluorinated allylic alcohols 1 corresponding esters 2 such as propionates, 3,3,3-trifluoropropionates and Boc-glycinates were synthesized. These compounds were rearranged to 2-substituted 4-fluoroalk-4-enecarboxylic acids 3 applying modified conditions of the [3,3]-sigmatropic Ireland-Claisen rearrangement. While a complete chirality transfer from C-3 of the allylic esters to C-2 of the carboxylic acids or 2-amino acids, respectively, occurred in rearrangements of the propionates and Boc-glycinates, racemic 2-(trifluoromethyl)alk-4-enecarboxylic acids were formed from the allylic trifluoropropionates. The configurational lability of the latter products is caused by the strongly acidic proton in α-position to the trifluoromethyl and the carboxyl groups under the basic rearrangement conditions.
Co-reporter:Nataliya A. Tolmachova;Igor I. Gerus;Sergey I. Vdovenko;Michael Essers;Rol Fröhlich and
European Journal of Organic Chemistry 2006 Volume 2006(Issue 20) pp:
Publication Date(Web):24 AUG 2006
DOI:10.1002/ejoc.200600408
A new practical method for the regioselective synthesis of the N-benzoyl-4-(polyfluoroalkyl)anilines 5a–g by thermal Diels–Alder cycloaddition of 5-substituted 3-(benzoylamino)-6-(polyfluoroalkyl)pyran-2-ones 1a–e with fluorostyrenes 2 and 7, acetylenes 8a–c or vinyl ethers 10 and 13a and 13b is described. In the case of the reactions of pyrone 1a with cyclic vinyl ethers 13a and 13b, the dihydrobenzenes 14a and 14b were obtained. Free 4-(polyfluoroalkyl)anilines 16a–c were smoothly formed in good yields by DBU-assisted benzoyl deprotection. (© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2006)
Co-reporter:Jens Oldendorf
European Journal of Organic Chemistry 2006 Volume 2006(Issue 19) pp:
Publication Date(Web):18 AUG 2006
DOI:10.1002/ejoc.200600456
Two diastereomeric enantiopure 4-fluoro-4,5-dihydroceramides with the natural D-erythro configuration at the 2- and 3-carbon atoms have been synthesized in an enantioselective eleven-step sequence. The key step of the synthesis was a diastereoselective asymmetric Sharpless dihydroxylation of ethyl trans-4-fluorooctadec-2-enoate (8) with AD-mix-β to afford the D-erythro arrangement. The diastereomers of the other enantiomeric series were synthesized analogously with the use of AD-mix-α. In all cases, the nitrogen heteroatom was introduced into the 2-position by regio- and stereoselective ring opening of cyclic sulfates 13 and 14 with azide. Staudinger reduction, acylation of the intermediary formed amino group, chromatographic separation of the diastereomers and chemoselective reduction of the ester functionality with sodium borohydride in the presence of methanol afforded both title compounds in an enantiopure form. (© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2006)
Co-reporter:Klaus Hegemann;Rol Fröhlich and
European Journal of Organic Chemistry 2004 Volume 2004(Issue 10) pp:
Publication Date(Web):27 APR 2004
DOI:10.1002/ejoc.200300783
Mixtures of endo,endo-9-oxabicyclo[4.2.1]nonane-2,5-diol (meso-2) and endo,endo-9-oxabicyclo[3.3.1]nonane-2,6-diol [(±)-3] were prepared from cycloocta-1,5-diene (1) upon treatment with peracids by transannular O-heterocyclization and subsequent saponification of the formed diol monoesters such as (±)-4 and (±)-5. The corresponding diacetates, meso-6 and (±)-7, were formed by acetylation of either meso-2 and (±)-3 or (±)-4 and (±)-5 with acetic anhydride/pyridine. These diacetates were enantioselectively hydrolyzed by microbial enzymes such as the lipases from Candida antarctica (CAL) or Candida rugosa (CRL). The corresponding enantiomers were formed by lipase-catalyzed acetylation of the diols meso-2 and (±)-3 with vinyl acetate. The skeletal isomers can also be separated in this way because the enantiopure monoacetates 4 were formed from the meso-compounds 2 or 6, while one enantiomer of the racemic diacetate (±)-7 [or the diol (±)-3] was transformed into the enantiopure diol 3 (or the enantiopure diacetate 7, respectively) via the corresponding enantiomers of the monoacetate 5. The other enantiomer remained untouched in both cases. The lipases reacted enantioselectively to give the R isomer. Cycloocta-1,5-diene (1) was also used to synthesize 2-oxa-6-thiatricyclo[3.3.1.13,7]decane-4,8-diol [(±)-11] in a four-step sequence. This racemic diol was also acetylated selectively (R isomer) with vinyl acetate and CRL. Reductive desulfuration of (±)-11 gave exo,exo-9-oxabicyclo[3.3.1]nonane-2,6-diol [(±)-12], which was acetylated selectively (S isomer) with CRL under the same conditions. The similarity in size and particularly in shape is responsible for the observed stereoselectivity of the lipases for the racemic endo,endo compounds (±)-3 and (±)-7 on the one hand and the exo,exo compound (±)-12 on the other hand. The absolute configuration and crystal packing of the products was determined by X-ray structural analysis. (© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2004)
Co-reporter:Sra Behr;Klaus Hegemann;Holger Schimanski;Rol Fröhlich and
European Journal of Organic Chemistry 2004 Volume 2004(Issue 18) pp:
Publication Date(Web):30 AUG 2004
DOI:10.1002/ejoc.200400219
The bislactones rac-tetrahydro-2,2′-bifuranyl-5,5′-dione (rac-12) and its diastereomer meso-25 were prepared from endo-5-hydroxy-9-oxabicyclo[4.2.1]nonan-2-one (endo-10) and endo-6-hydroxy-9-oxabicyclo[3.3.1]nonan-2-one (endo-11) or exo-5-hydroxy-9-oxabicyclo[4.2.1]nonan-2-one (exo-23), respectively, under the conditions of a Baeyer−Villiger oxidation with trifluoroperacetic acid. The latter compounds were obtained by O-heterocylization of cis,cis-cycloocta-1,5-diene (1) by either reaction with peracids followed by hydrolysis and Jones oxidation or ruthenium tetraoxide oxidation, respectively. The optically active bislactone (R,R)-(−)-12 was prepared in a similar manner from (1S,5R,6R)-(+)-10 and (1R,5R,6R)-(+)-11, which, in turn, were obtained by lipase-catalyzed asymmetric acetylation of the corresponding diols meso-2 and rac-3 and subsequent Jones oxidation of the formed hydroxy esters (1S,2S,5R,6R)-(+)-4 and (1R,2R,5R,6R)-(+)-5. Since the regioisomeric hydroxy-9-oxabicyclo[4.2.1]- and -[3.3.1]nonan-2-ones (1S,5R,6R)-(+)-10 and (1R,5R,6R)-(+)-11 yielded the same bislactone [(R,R)-(−)-12] it is presumed that the sequence proceeds via open-chain intermediates. Applying this strategy to the enantiopure acetoxy ketones (1S,5R,6R)-(+)-8 and (1R,5R,6R)-(+)-9, followed by Kolbe electrolysis of the formed (R,R)-5-acetoxy-7-carboxyheptan-4-olide [(R,R)-27], (R,R)-5-acetoxydecan-4-olide [(R,R)-29] was accessible in a five-step synthesis. The absolute configuration of (+)-8 was determined by X-ray analysis of the dithiepane derivative 14. (© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2004)
Co-reporter:Thomas C. Rosen Dr.;Shinichi Yoshida Dr.;Kenneth L. Kirk Dr. Dr.
ChemBioChem 2004 Volume 5(Issue 8) pp:
Publication Date(Web):2 AUG 2004
DOI:10.1002/cbic.200400053
Location makes a difference. Fluorinated analogues of the antidepressive tranylcypromine (see structures) are much stronger inhibitors of different monoamine oxidases such as microbial tyramine oxidase or human liver mitochondrial-outer-membrane monoamine oxidases A and B. Regiochemistry, as well as relative and absolute stereochemistry of the fluorine substituent at the cyclopropane ring, has a strong influence on the effectiveness of inhibition and on the physicochemical data, such as pKa values, permeability, and log D values.
Co-reporter:Günter Haufe;Dörthe Wölker
European Journal of Organic Chemistry 2003 Volume 2003(Issue 11) pp:
Publication Date(Web):19 MAY 2003
DOI:10.1002/ejoc.200200647
The biotransformation of tricyclo[2.2.1.02,6]hept-3-yl N-phenylcarbamate (8) by a standard procedure using Beauveria bassiana gave a 7:1 mixture of optically active exo,exo- and exo,endo-5-hydroxytricyclo[2.2.1.02,6]hept-3-yl N-phenylcarbamates 15 and 16 in 19% isolated yield. No ring opening of the three-membered ring was observed. Substitution with a fluorine atom at the 5-endo- or 5-exo-position prevented hydroxylation of any alicyclic position of the molecules, p-hydroxylation of the aromatic ring occurring to a small extent instead. (© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2003)
Co-reporter:Günter Haufe;Stefan Bruns
Advanced Synthesis & Catalysis 2002 Volume 344(Issue 2) pp:
Publication Date(Web):27 APR 2002
DOI:10.1002/1615-4169(200202)344:2<165::AID-ADSC165>3.0.CO;2-1
The asymmetric ring opening of five meso- and three racemic epoxides with different fluorinating reagents in the presence of stoichiometric or slightly sub-stoichiometric amounts of Jacobsen's enantiopure (salen)chromium chloride complex A gave the corresponding optically active vicinal fluorohydrins. Silver fluoride was used as one of the fluoride sources either in the presence of Bu4N+H2F3− in diethyl ether or in acetonitrile. The latter reactions starting from cyclohexene oxide (1) showed maximum 72% ee in the formed fluorohydrin 2 isolated in 90% yield. From other meso-epoxides such as cyclopentene oxide and cycloheptene oxide the corresponding fluorohydrins were isolated in 80% and 82% yield with 65% and 62% ee, respectively. In case of ring opening under similar conditions of the racemic styrene oxide or phenyl glycidyl ether 83% and 75% of the fluorohydrins with fluorine in the primary position were isolated with 74% ee and 65% ee, respectively. Tetrahydronaphthalene oxide yielded a 2:1 mixture of trans- (23% ee) and cis-2-fluoro-3,4-benzocyclohexenol (2% ee) suggesting competing SN2 and SN1 type ring openings. Other epoxides such as cyclooctene oxide, cis-stilbene oxide and α-methylstyrene oxide did not react or gave the fluorohydrins with very small enantiomeric excess.
Co-reporter:Michael Essers and Günter Haufe
Organic & Biomolecular Chemistry 2002 (Issue 23) pp:2719-2728
Publication Date(Web):24 Oct 2002
DOI:10.1039/B208001J
Four fluorinated p-benzoquinones (2) have been reacted with Dane's diene (1) in Diels–Alder reactions and the formed fluorinated D-homosteroids were characterized. The number of products, their stereochemistry and stability depends on the fluorine substitution pattern of the corresponding fluorinated p-benzoquinones. If the p-benzoquinone (2) contains an unfluorinated double bond, this bond reacts faster with diene 1 yielding endo-products selectively. In contrast, [4+2]cycloadditions with 2,6-difluoro (2c) and 2,3,5,6-tetrafluorobenzoquinone (2d) gave the products with exo-orientation of the carbonyl part preferably.
Co-reporter:Günter Haufe, Thomas C. Rosen, Oliver G.J. Meyer, R. Fröhlich, Kari Rissanen
Journal of Fluorine Chemistry 2002 Volume 114(Issue 2) pp:189-198
Publication Date(Web):28 April 2002
DOI:10.1016/S0022-1139(02)00040-4
Monofluorinated cyclopropanecarboxylates are available in racemic or optically active form by transition metal-catalyzed reactions of vinylfluorides with diazoacetates. From α-fluorostyrene and tert-butyl diazoacetate in the presence of 2 mol% of an enantiopure bis(oxazoline) copper complex, a 81:19 mixture of tert-butyl trans- and cis-2-fluoro-2-phenylcyclopropanecarboxylates was obtained with high enantiomeric excess (ee) of 93 or 89%, respectively. The corresponding racemic ethylesters were used as starting materials for the synthesis of carboxamides, of the cis- and trans-isomers of analogues of tranylcypromine, an anti-depressive drug and several of its homologous fluorinated cyclopropylmethyl and cyclopropylethyl amines. Corresponding enantiopure cyclopropylmethanols and several of their derivatives were synthesized also. Solid state structures of a selection of these compounds were examined by X-ray crystallography. Particularly, the cis-configurated fluorinated phenylcyclopropane derivatives showed extremely close intermolecular CH⋯FC contacts. The shortest of such distances (2.17 Å) was found in the N-(4-bromophenyl)carbamate of (1S,2R)-(2-fluoro-2-phenylcyclopropyl)methanol.Graphic
Co-reporter:Thomas C. Rosen, Günter Haufe
Tetrahedron: Asymmetry 2002 Volume 13(Issue 13) pp:1397-1405
Publication Date(Web):19 July 2002
DOI:10.1016/S0957-4166(02)00331-2
All four stereoisomers of (2-fluoro-2-phenlycyclopropyl)methanol were synthesized using Amano PS lipase-catalyzed acylation and transesterification as key steps. Copper(I)-catalyzed cyclopropanation of α-fluoro styrene with ethyl diazoacetate and chromatographic separation gave diastereomerically pure cyclopropane carboxylates, which were then reduced with LiAlH4. Whereas the enzymatic acylation gave low selectivities for trans-configured alcohols (E=13) using different acyl donors, the corresponding cis-diastereomers were obtained with high enantiomeric excesses (E >200). Additionally, kinetic resolution of different racemic esters of (2-fluoro-2-phenlycyclopropyl)methanol was achieved by lipase-catalyzed transesterification employing Amano PS in the presence of ethanol and the selectivities from this process were found to be comparable to those observed in the enzymatic acylation. The absolute configurations of the enantiomers were confirmed by X-ray structural analysis of the corresponding enantiopure p-bromophenyl carbamates. In addition, the stereochemistry of the product from the asymmetric cyclopropanation of α-fluoro styrene with a chiral bis(oxazoline) copper(I) catalyst was similarly determined.Graphic(1R,2R)-(2-Fluoro-2-phenylcyclopropyl)methanolC10H11FOE.e.=98% by GC on chiral Beta-Dex™ 120 (isotherme, 140°C)[α]25D=+51.3 (c 1.0 in CHCl3)Source of chirality: Amano PS-catalyzed deracemizationAbsolute configuration: 1R,2R(1S,2S)-(2-Fluoro-2-phenylcyclopropyl)methyl acetateC12H13FO2E.e.=66% by GC on chiral Beta-Dex™ 120 (isotherme, 140°C) after hydrolysis with KOH/MeOH[α]25D=−36.2 (c 1.0 in CHCl3)Source of chirality: Amano PS-catalyzed acetylationAbsolute configuration: 1S,2S(1S,2S)-(2-Fluoro-2-phenylcyclopropyl)methyl chloroacetateC12H12ClFO2E.e.=47% by GC on chiral Beta-Dex™ 120 (isotherme, 140°C) after hydrolysis with KOH/MeOH[α]25D=−22.2 (c 1.0 in CHCl3)Source of chirality: Amano PS lipase-catalyzed acetylationAbsolute configuration: 1S,2S(1S,2S)-(2-Fluoro-2-phenylcyclopropyl)methyl propionateC13H15FO2E.e.=69% by GC on chiral Beta-Dex™ 120 (isotherme, 140°C) after hydrolysis with KOH/MeOH[α]25D=−35.5 (c 1.0 in CHCl3)Source of chirality: Amano PS lipase-catalyzed propyrylationAbsolute configuration: 1S,2S(1R,2R)-(2-Fluoro-2-phenylcyclopropyl)methyl (4-bromophenyl)carbamateC17H15BrFNO2E.e. >99%[α]25D=+17.4 (c 1.0 in CHCl3)Source of chirality: synthesis from (1R,2R)-(2-fluoro-2-phenylcyclopropyl)methanolAbsolute configuration: 1R,2R(1S,2R)-(2-Fluoro-2-phenylcyclopropyl)methanolC10H11FOE.e.=90% by GC on chiral Beta-Dex™ 120 (isotherme, 135°C)[α]25D=+20.8 (c 1.0 in CHCl3)Source of chirality: Amano PS-catalyzed deracemizationAbsolute configuration: 1S,2R(1R,2S)-(2-Fluoro-2-phenylcyclopropyl)methyl acetateC12H13FO2E.e.=99% by GC on chiral Beta-Dex™ 120 (isotherme, 135°C) after hydrolysis with KOH/MeOH[α]25D=−19.2 (c 1.0 in CHCl3)Source of chirality: Amano PS-catalyzed acetylationAbsolute configuration: 1R,2S(1R,2S)-(2-Fluoro-2-phenylcyclopropyl)methyl chloroacetateC12H12ClFO2E.e.=80% by GC on chiral Beta-Dex™ 120 (isotherme, 135°C) after hydrolysis with KOH/MeOH[α]25D=−17.5 (c 1.0 in CHCl3)Source of chirality: Amano PS lipase-catalyzed acetylationAbsolute configuration: 1R,2S(1R,2S)-(2-Fluoro-2-phenylcyclopropyl)methyl propionateC13H15FO2E.e.=97% by GC on chiral Beta-Dex™ 120 (isotherme, 135°C) after hydrolysis with KOH/MeOH[α]25D=−22.2 (c 1.0 in CHCl3)Source of chirality: Amano PS lipase-catalyzed propyrylationAbsolute configuration: 1R,2S(1S,2R)-(2-Fluoro-2-phenylcyclopropyl)methyl (4-bromophenyl)carbamateC17H15BrFNO2E.e. >98%[α]25D=+40.7 (c 1.0 in CHCl3)Source of chirality: synthesis from (1S,2R)-(2-fluoro-2-phenylcyclopropyl)methanolAbsolute configuration: 1S,2R(−)-Menthyl (1S,2R)-2-fluoro-2-phenylcyclopropane carboxylateC20H27FO2D.e.=88% by 19F NMR spectroscopy[α]25D=−25.9 (c 1.0 in CHCl3)Source of chirality: synthesis from (1S,2R)-(2-fluoro-2-phenylcyclopropyl)methanolAbsolute configuration: 1S,2R
Co-reporter:Günter Haufe
Advanced Synthesis & Catalysis 2001 Volume 343(Issue 3) pp:
Publication Date(Web):19 MAR 2001
DOI:10.1002/1615-4169(20010330)343:3<311::AID-ADSC311>3.0.CO;2-K
Co-reporter:Thomas Ernet, Andreas H. Maulitz, Ernst-Ulrich Würthwein and Günter Haufe
Organic & Biomolecular Chemistry 2001 (Issue 16) pp:1929-1938
Publication Date(Web):02 Aug 2001
DOI:10.1039/B102684B
Vinyl fluorides such as α- and β-fluorostyrenes 2 and 3 are poor dienophiles for Diels–Alder reactions. Under thermal conditions these compounds do not react with usual dienes, but with the highly reactive 1,3-diphenylisobenzofuran (4) to give mixtures of the corresponding endo- and exo-products. Kinetic measurements show that the fluorostyrenes 2 and 3 are less reactive than the parent styrene (1a). Additional electron withdrawing as well as electron donating substituents (p-Cl, p-F, m-Me) on the phenyl ring of 2 and 3 slightly accelerate the reaction rate by a maximum factor of 4. DFT calculations (UB3LYP/6-31G(d)) of activation energies for furan and isobenzofuran as model dienes reflect the order of the reaction rates of the kinetic measurements. The charge difference in the double bond carbon atoms of the different dienophiles pre-determines the activation energies for the endo- and exo-transition states. The optimised structures of the transition states show that the reactions are concerted but asynchronous without biradical character. This asynchrony is determined by the differences in the pz orbital coefficients of the double bond carbon atoms of the fluorostyrenes. The frontier molecular orbitals reveal that all the reactions are cycloadditions with normal electron demand. The calculated small endo/exo preferences are in qualitative agreement with the experimental values. In the transition states of the reactions of 2a and
(E)-3a where the fluorine atom and the oxygen atom of furan are syn-orientated with respect to the reaction center, electrostatic repulsion determines the endo/exo selectivity. Semiempirical calculations show that the reactions of 4 are more exothermic with respect to the parent furan and more endothermic with respect to isobenzofuran. However, regarding relative reactivity and diastereoselectivity, the semiempirical methods are less reliable in comparison to either experiment or DFT.
Co-reporter:Günter Haufe;Annegret Burchardt
European Journal of Organic Chemistry 2001 Volume 2001(Issue 23) pp:
Publication Date(Web):7 NOV 2001
DOI:10.1002/1099-0690(200112)2001:23<4501::AID-EJOC4501>3.0.CO;2-J
The synthesis of racemic 2-fluoro-3-hexadecyloxy-2-methylprop-1-yl 2′-(trimethylammonio)ethyl phosphate (10), a fluorinated analogue of the anticancer active and blood platelet activating ether lipids 8 and 9, has been achieved in a six-step sequence from methallyl alcohol (11). Etherification of 11 with hexadecyl bromide gave allylic ether 12, bromofluorination of which afforded the bromo-substituted fluoride 13, which was subsequently transformed into the acetate 14. Hydrolysis of 14 gave the key intermediate 2-fluoro-2-(hexadecyloxymethyl)propanol (15), which was attached to the phosphocholine residue in the final step. Enzyme-catalyzed deracemization of the fluorohydrin 15 by acetylation with several lipases gave optically active compounds 14 and 15, with a maximum ee of 61% for 15 with Candida antarctica lipase catalysis after 74% conversion. An enantioselective synthesis of 10, based on a planned eight-step synthesis starting from α-methylstyrene, failed. The anticancer activity of racemic 10 has been observed in an in vivo model of methylcholanthrene-induced mouse fibrosarcoma.
Co-reporter:Stephan Otten, Roland Fröhlich, Günter Haufe
Tetrahedron: Asymmetry 2001 Volume 12(Issue 1) pp:175
Publication Date(Web):5 February 2001
DOI:10.1016/S0957-4166(01)00007-6
Co-reporter:Klaus W. Laue;Stefan Kröger;Elina Wegelius
European Journal of Organic Chemistry 2000 Volume 2000(Issue 22) pp:
Publication Date(Web):19 OCT 2000
DOI:10.1002/1099-0690(200011)2000:22<3737::AID-EJOC3737>3.0.CO;2-A
(+)-(S)-2-Amino-4-fluorobutanoic acid (5a) (> 96% ee), its α-methylated derivative (+)-(S)-5b (85% ee), and (−)-(S)-2-amino-4-fluoro-4-pentenoic acid (10) (81% ee) were synthesized by diastereoselective alkylation in the presence of LDA at low temperatures. Alkylation of (+)-(R,R,R)-2-hydroxy-3-pinanone based imines of glycine tert-butyl ester 1a or alanine isopropyl ester 1b with 1-bromo-2-fluoroethane (2) or 3-bromo-2-fluoropropene (7), respectively, followed by stepwise deprotection was used. The selectivity of the alkylation increased by lithium/magnesium exchange or for 1b also by addition of DMPU. Differences in the reactivity of enolate alkylations of enantiomerically pure or racemic Schiff base 1a with 2 or 7, respectively, can be explained by the formation of structurally different aggregates of the enolates in solution, caused by diastereomeric interactions between enantiomers in the transition state of alkylation.
Co-reporter:Martina Runge;David O'Hagan
Journal of Polymer Science Part A: Polymer Chemistry 2000 Volume 38(Issue 11) pp:2004-2012
Publication Date(Web):3 MAY 2000
DOI:10.1002/(SICI)1099-0518(20000601)38:11<2004::AID-POLA90>3.0.CO;2-T
Lipase-catalyzed ring-opening polymerization of 10-fluorodecan-9-olide (1a), 11-fluoroundecan-10-olide (1b), 12-fluorododecan-11-olide (1c) and 14-fluorotetradecan-13-olide (1d) gave optically active products with Mw of 3.000 to 8.000, while 10-fluoroundecan-11-olide (3a) gave an optically inactive polymer with Mw = 11.000. On the other hand, Candida antarctica lipase-catalyzed polymerization of 10- to 14-membered ω-fluoro-(ω-1)-hydroxyalcanoic acids gave optically inactive oligomers with Mw of 3.000 to 11.000 in the presence of molecular sieves, while reactions without molecular sieves gave oligomers with lower molecular weight, but with small optical rotations. 9-Fluoro-10-hydroxydecanoic acid, on the other hand, gave an optically inactive polyester with Mw = 7400. © 2000 John Wiley & Sons, Inc. J Polym Sci A: Polym Chem 38: 2004–2012, 2000
Co-reporter:Frank Tranel, Günter Haufe
Tetrahedron: Asymmetry 2000 Volume 11(Issue 4) pp:889-896
Publication Date(Web):10 March 2000
DOI:10.1016/S0957-4166(99)00572-8
Kinetic resolutions of alkyl (±)-2-fluorodecanoates by lipase-catalyzed hydrolysis and of (±)-2-fluorodecanoic acid by lipase-catalyzed esterification are described for the first time.
Co-reporter:Stefan Kröger
European Journal of Organic Chemistry 1997 Volume 1997(Issue 6) pp:
Publication Date(Web):25 JAN 2006
DOI:10.1002/jlac.199719970623
(R)-(−)-2-Amino-4-fluorobutyric acid (4c) (32% ee) and six of its homologues 9 have been synthesized by diastereoselective alkylation (54–72% yield) of (R)-(+)-camphor-based glycine ester imines 1 with 1-bromo-2-fluoro-alkanes 6, at low temperature, followed by deprotection. Similarly 1-bromo-3-fluoropropane was used for the preparation of (R)-(−)-5-fluornorvaline (4d) (42% ee). With 1-bromo-2-fluoropropane (6a) and its homologues (prepared by bromofluorination of 1-alkenes) partial resolution occurs in the alkylation step, yielding mixtures of four diastereomers. Using (R)-1-bromo-2-fluoro-4-methylpentane two diastereomeric alkylation products are formed (58% de). The overall yield of the three-step procedure varied from 10% to 32%.
Co-reporter:Olav Goj, Annegret Burchardt, Günter Haufe
Tetrahedron: Asymmetry 1997 Volume 8(Issue 3) pp:399-408
Publication Date(Web):6 February 1997
DOI:10.1016/S0957-4166(96)00501-0
Kinetic resolutions of (±)-1-acetoxy-2-fluoro-2-phenylalkanes 1 by enzymatic hydrolysis and of (±)-2-fluoro-2-phenylpropanol 2a by lipase-catalyzed acetylation are described for the first time. Hydrolysis of (±)-1 with lipase Amano PS (Pseudomonas cepacia) provided both the optically active acetates (−)-1 and the corresponding primary alcohols (−)-2 with high enantiomeric excess. (R)-enantiopreference was observed for the acetylation of (±)-2-fluoro-2-phenylpropanol 2a which occurred with higher enantioselectivity and faster conversion compared to the unfluorinated parent compound (±)-2-phenylpropanol 3.In addition to hydrolysis of acetates also the acetylation of 2-fluoro-2-phenyl-propanol is used for kinetic deracemization using Pseudomonas cepacia lipase Amano PS.
Co-reporter:T. Borisova, N. Pozdnyakova, E. Shaitanova, I. Gerus, M. Dudarenko, G. Haufe, V. Kukhar
Bioorganic & Medicinal Chemistry (15 January 2017) Volume 25(Issue 2) pp:
Publication Date(Web):15 January 2017
DOI:10.1016/j.bmc.2016.11.052
•Properties of new synthesized fluorinated analogues of GABA were analyzed.•The analogues did not influence the ambient level of [3H]GABA in nerve terminals.•The analogues decreased exocytotic release of [3H]GABA from nerve terminals.•Comparative analysis of the effects of the analogues and pregabalin was performed.Recently, we have shown that new fluorinated analogues of γ-aminobutyric acid (GABA), bioisosters of pregabalin (β-i-Bu-GABA), i.e. β-polyfluoroalkyl-GABAs (FGABAs), with substituents: β-CF3-β-OH (1), β-CF3 (2); β-CF2CF2H (3), are able to increase the initial rate of [3H]GABA uptake by isolated rat brain nerve terminals (synaptosomes), and this effect is higher than that of pregabalin. So, synthesized FGABAs are structural but not functional analogues of GABA. Herein, we assessed the effects of synthesized FGABAs (100 μM) on the ambient level and exocytotic release of [3H]GABA in nerve terminals and compared with those of pregabalin (100 μM). It was shown that FGABAs 1–3 did not influence the ambient level of [3H]GABA in the synaptosomal preparations, and this parameter was also not altered by pregabalin. During blockage of GABA transporters GAT1 by specific inhibitor NO-711, FGABAs and pregabalin also did not change ambient [3H]GABA in synaptosomal preparations. Exocytotic release of [3H]GABA from synaptosomes decreased in the presence of FGABAs 1–3 and pregabalin, and the effects of FGABAs 1 & 3 were more significant than those of FGABAs 2 and pregabalin. FGABAs 1–3/pregabalin-induced decrease in exocytotic release of [3H]GABA from synaptosomes was not a result of changes in the potential of the plasma membrane. Therefore, new synthesized FGABAs 1 & 3 were able to decrease exocytotic release of [3H]GABA from nerve terminals more effectively in comparison to pregabalin. Absence of unspecific side effects of FGABAs 1 & 3 on the membrane potential makes these compounds perspective for medical application.
Co-reporter:Ivan S. Kondratov, Ivan G. Logvinenko, Nataliya A. Tolmachova, Roman N. Morev, Maria A. Kliachyna, Florian Clausen, Constantin G. Daniliuc and Günter Haufe
Organic & Biomolecular Chemistry 2017 - vol. 15(Issue 3) pp:NaN679-679
Publication Date(Web):2016/12/06
DOI:10.1039/C6OB02436J
2-Amino-2-(trifluoromethoxy)butanoic acid (O-trifluoromethyl homoserine) was synthesized as a racemate and in both enantiomeric forms. The measured pKa and logD values establish the compound as a promising analogue of natural aliphatic amino acids.
Co-reporter:Isabella Hyla-Kryspin, Stefan Grimme, Svenja Hruschka and Günter Haufe
Organic & Biomolecular Chemistry 2008 - vol. 6(Issue 22) pp:NaN4175-4175
Publication Date(Web):2008/09/22
DOI:10.1039/B810108F
Fluorine substituents in organic molecules do dramatically influence the electronic structure of neighbouring functional groups and the conformation of molecules. Hence the presence of fluorine in a compound changes its chemical reactivity and biological activity. On the basis of MP2 and SCS-MP2 calculations, we discuss the conformational preferences and basicity of the diastereoisomeric 2-fluorocyclopropylamines (cis-2 and trans-2) in comparison to those of cyclopropylamine (1) and 2-fluoroethylamine (3). 1 and 2 are viewed as model compounds for the antidepressant drug tranylcypromine (trans-2-phenylcyclopropylamine, 1′a) and its fluorinated derivatives 2′. The potential energy profile for the rotation of the amino group in cis-2 differs from that of trans-2 and 1 which have very similar rotational curves. For 2 the global minimum conformer is trans-2a and the lowest energy cis-conformer 2c is less stable by 2.57 kcal mol−1. The calculated enthalpy differences between the conformers gauche-1b and s-trans-1a (2.0 kcal mol−1) as well as between gauche-3b and gauche-4a (0.2 kcal mol−1) agree well with the available experimental data of 2.0 kcal mol−1 and 0.1 ± 0.3 kcal mol−1, respectively. The calculated gas phase proton affinities (PA) of 1 (217.6 kcal mol−1), cis-2c (215.6 kcal mol−1), and trans-2a (209.3 kcal mol−1) follow the trends of the pKa values measured in solution for the diastereomeric 2-phenylcyclopropylamines 1′a and 1′b and their fluorinated derivatives cis-2′ and trans-2′. It is shown that the conformational preferences and basicity of the investigated molecules are due to stereoelectronic effects from hyperconjugative interactions which lead to different local charge distributions and different hybridization of the nitrogen lone-pair. The basicity of gauche-3a (PA = 215.3 kcal mol−1) and anti-3b (PA = 210.1 kcal mol−1) is controlled by the charge of the nitrogen atom, while that of cis-2c and trans-2a is overlap controlled as a result of different hybridization of the nitrogen lone-pair [sp4.34 (cis-2c), sp4.07 (trans-2a)].
Co-reporter:Dirk Ulbrich, Constantin G. Daniliuc and Günter Haufe
Organic & Biomolecular Chemistry 2016 - vol. 14(Issue 9) pp:NaN2767-2767
Publication Date(Web):2016/01/29
DOI:10.1039/C6OB00131A
Intending to synthesize ω,ω-difluoroalkyl amino acid derivatives by oxidative desulfurization–fluorination reactions of suitable arylthio-2-phthalimido butanoates and pentanoates, in addition to small amounts of the target products, mainly α,ω-polyfluorinated amino acid derivatives were formed by additional sulfur-assisted α-fluorination. This novel structural motif was verified spectroscopically as well as by X-ray analysis. A plausible mechanism of formation is suggested. Using a different approach, δ,δ-difluoronorvaline hydrochloride was synthesized with at least 36% enantiomeric excess via deoxofluorination of the corresponding aldehyde.
Co-reporter:Verena Hugenberg, Roland Fröhlich and Günter Haufe
Organic & Biomolecular Chemistry 2010 - vol. 8(Issue 24) pp:NaN5691-5691
Publication Date(Web):2010/10/21
DOI:10.1039/C0OB00560F
An oxidative desulfurization–fluorination protocol has been used to synthesize (2S)-2-(difluoromethyl)-N-tosylpyrrolidine (6a) and (2S)-2-(trifluoromethyl)-N-tosylpyrrolidine (7a) from the (2S)-prolinol-derived (2S)-2-(4-chlorophenylthiomethyl)-N-tosylpyrrolidine (9) or (2S)-2-(dithian-2-yl)-N-tosylpyrrolidine (5). Efforts to prepare 3,3-difluoroalanine similarly from an N-protected S-aryl-cysteine ester 17 gave only traces of the target compound 18. Instead, an unique N-(α,α-difluorobenzyl)-N-α′,α′-dibromoglycine ester 19 was formed by an unprecedented sequence of reaction steps. A plausible mechanism is suggested involving a sulfur-assisted deoxygenation-difluorination of an imino oxygen and a haloform reaction like carbon–carbon bond fission as key-steps. Efforts to prepare (2S)-2-(fluoromethyl)-N-tosylpyrrolidine (12) from (2S)-N-tosylprolinol (3) by treatment with Fluolead™ (1-tert-butyl-4-trifluorosulfanyl-3,5-dimethylbenzene) gave only 5% of the target compound, but 95% of (3R)-3-fluoro-N-tosylpiperidine (11a) by ring enlargement.
Co-reporter:Ionel Humelnicu, Ernst-Ulrich Würthwein and Günter Haufe
Organic & Biomolecular Chemistry 2012 - vol. 10(Issue 10) pp:NaN2093-2093
Publication Date(Web):2011/12/07
DOI:10.1039/C2OB06492H
Quantum chemical calculations (DFT, SCS-MP2) show that the relative energies of the four principal alanine conformations are only marginally altered by the introduction of a single fluorine substituent into the methyl group. The fluorinegauche effect and attractive interactions of fluorine to the O–H or N–H moieties (formation of hydrogen bridges) do stabilize particular conformers of 3-fluoroalanine. This is true for the neutral molecule both in the gas phase and in aqueous solution (CPCM-model), but also for the zwitterionic forms and the conformers of the related carboxylate ions and also for the respective ammonium ions in aqueous solution. In water (CPCM calculations), the zwitterion is almost equal in energy to the most stable conformer of the neutral 3-fluoroalanine. Compared to alanine the atomic charges of the amino group and the carboxyl function of 3-fluoroalanine are not significantly influenced by the fluorine at C3, which relates to the fact that both experimental pKa values are almost equal for alanine and 3-fluoroalanine.
Co-reporter:Ivan S. Kondratov, Violetta G. Dolovanyuk, Nataliya A. Tolmachova, Igor I. Gerus, Klaus Bergander, Roland Fröhlich and Günter Haufe
Organic & Biomolecular Chemistry 2012 - vol. 10(Issue 44) pp:NaN8785-8785
Publication Date(Web):2012/09/04
DOI:10.1039/C2OB26176F
The hitherto unreported reactions of β-alkoxyvinyl polyfluoroalkyl ketones with ethyl isocyanoacetate and equimolar amounts of potassium-tert-butoxide proceeded mainly in the β-position of the α,β-unsaturated ketones in cases of α-nonsubstituted 1a–e and α-methyl substituted ketones 1g–j. Other α- or β-substituted ketones 1f,k–o gave mainly products 4 of initial attack at the carbonyl carbon. Depending on the solvent, the major products of β-attack do exist in different tautomeric forms. Generally the open-chain enol tautomers 5 predominate in the polar DMSO-d6, while the cyclic γ-hemiaminals 8 are the major tautomers in the less polar CDCl3. Acid treatment of the latter compounds 8 led to the hitherto unknown ethyl 5-polyfluoroalkyl-pyrrole-2-carboxylates 11 by elimination of formic acid. Catalytic hydrogenation of pyrrole 11a was used for the synthesis of earlier unknown 5-trifluoromethyl proline 16.

![1,3-Propanediol, 2-amino-2-[2-[4-(8-fluorooctyl)phenyl]ethyl]-](http://img.cochemist.com/ccimg/801300/801289-27-0.png)
![1,3-Propanediol, 2-amino-2-[2-[4-(8-fluorooctyl)phenyl]ethyl]-](http://img.cochemist.com/ccimg/801300/801289-27-0_b.png)
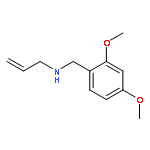
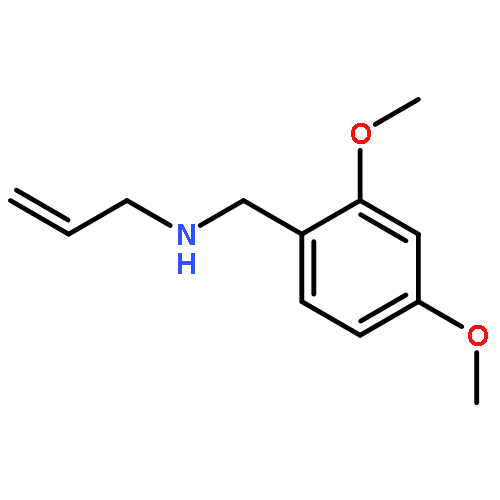
![2(1H)-Pyridinone, 3-fluoro-1-[(4-fluorophenyl)methyl]-5,6-dihydro-](http://img.cochemist.com/ccimg/664400/664342-31-8.png)
![2(1H)-Pyridinone, 3-fluoro-1-[(4-fluorophenyl)methyl]-5,6-dihydro-](http://img.cochemist.com/ccimg/664400/664342-31-8_b.png)
![2(1H)-PYRIDINONE, 3-FLUORO-5,6-DIHYDRO-1-[(4-METHOXYPHENYL)METHYL]-](http://img.cochemist.com/ccimg/664400/664342-30-7.png)
![2(1H)-PYRIDINONE, 3-FLUORO-5,6-DIHYDRO-1-[(4-METHOXYPHENYL)METHYL]-](http://img.cochemist.com/ccimg/664400/664342-30-7_b.png)
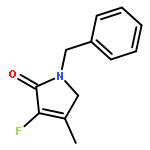
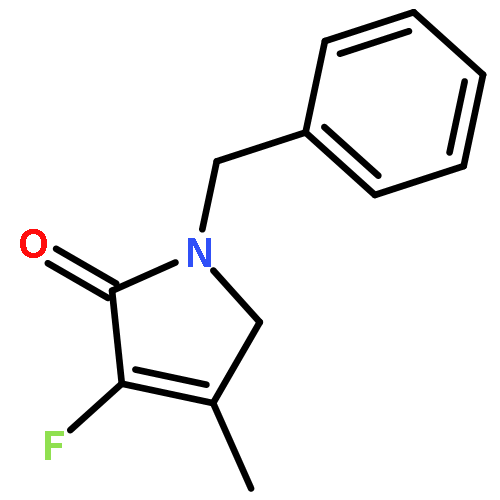
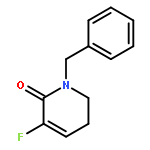
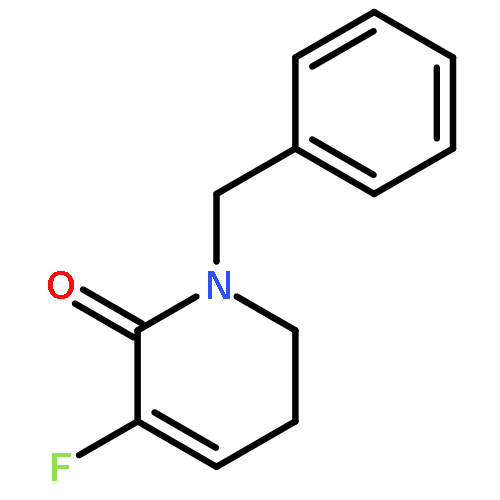
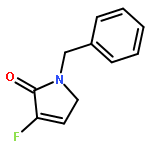
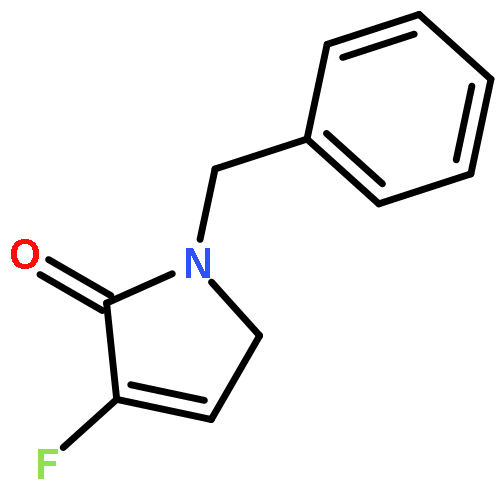
![2-Propenamide, N-3-butenyl-2-fluoro-N-[(4-fluorophenyl)methyl]-](http://img.cochemist.com/ccimg/664400/664342-26-1.png)
![2-Propenamide, N-3-butenyl-2-fluoro-N-[(4-fluorophenyl)methyl]-](http://img.cochemist.com/ccimg/664400/664342-26-1_b.png)
![2-PROPENAMIDE, N-3-BUTENYL-2-FLUORO-N-[(4-METHOXYPHENYL)METHYL]-](http://img.cochemist.com/ccimg/664400/664342-25-0.png)
![2-PROPENAMIDE, N-3-BUTENYL-2-FLUORO-N-[(4-METHOXYPHENYL)METHYL]-](http://img.cochemist.com/ccimg/664400/664342-25-0_b.png)
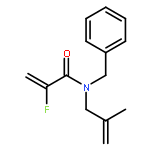
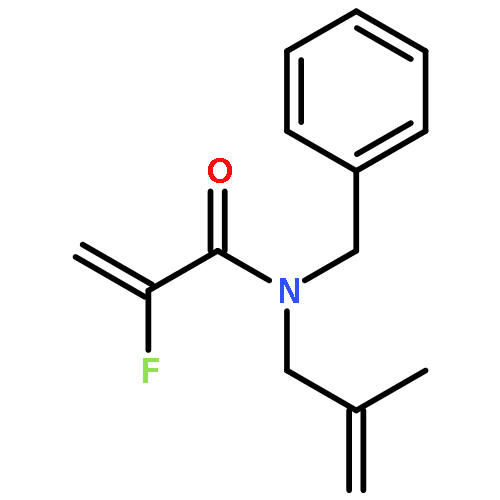
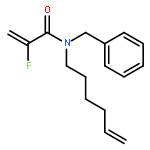
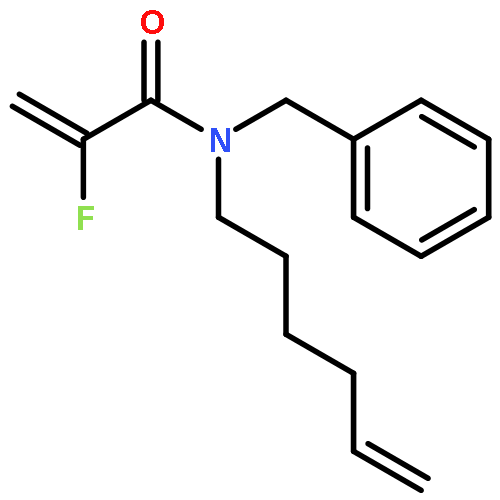
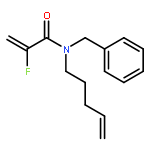
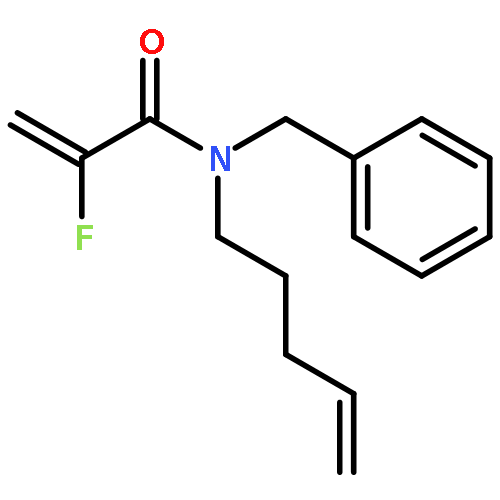
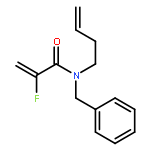
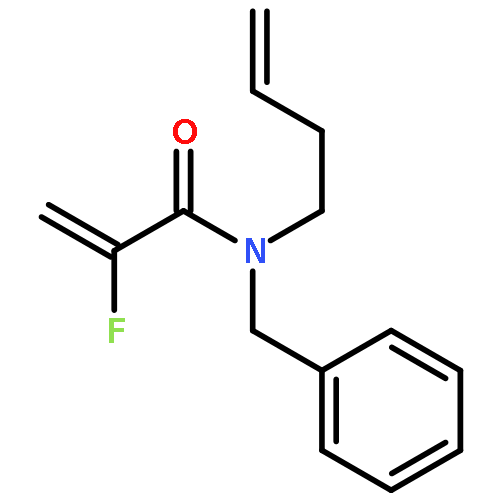
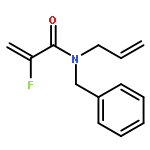
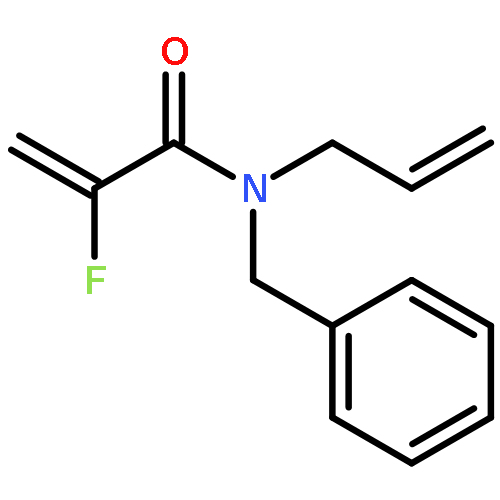
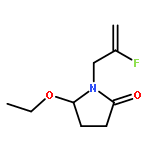
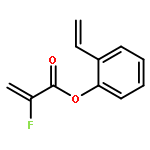
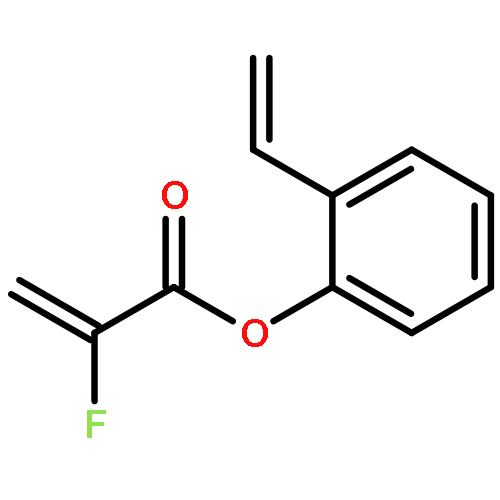
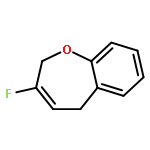
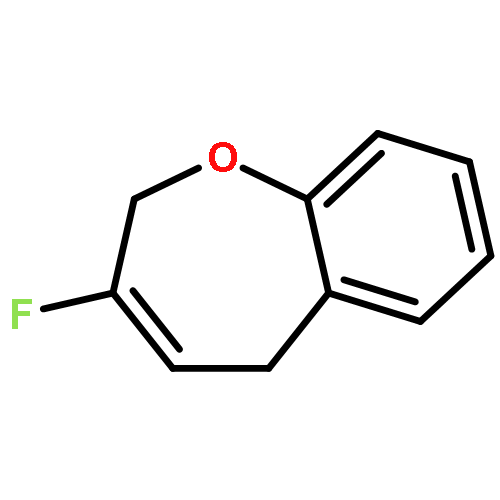
![BENZENE, 1-[(2-FLUORO-2-PROPENYL)OXY]-2-(2-PROPENYL)-](http://img.cochemist.com/ccimg/607000/606926-71-0.png)
![BENZENE, 1-[(2-FLUORO-2-PROPENYL)OXY]-2-(2-PROPENYL)-](http://img.cochemist.com/ccimg/607000/606926-71-0_b.png)
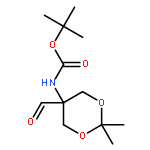

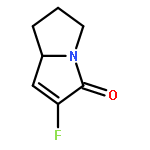
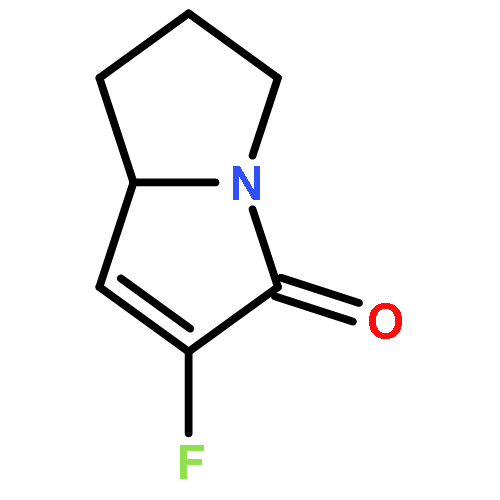
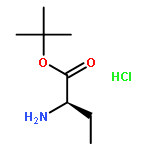
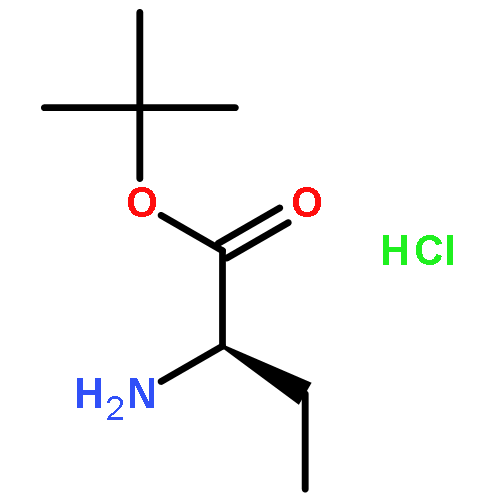
![2-(acetylamino)-2-[2-(4-octylphenyl)-2-oxo-ethyl]propanedioic acid diethyl ester](http://img.cochemist.com/ccimg/268600/268557-49-9.png)
![2-(acetylamino)-2-[2-(4-octylphenyl)-2-oxo-ethyl]propanedioic acid diethyl ester](http://img.cochemist.com/ccimg/268600/268557-49-9_b.png)
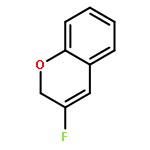
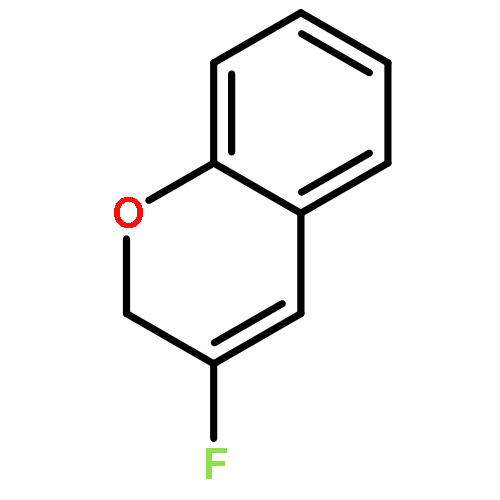
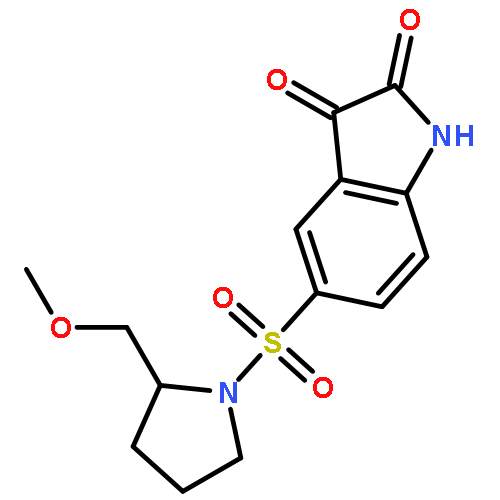
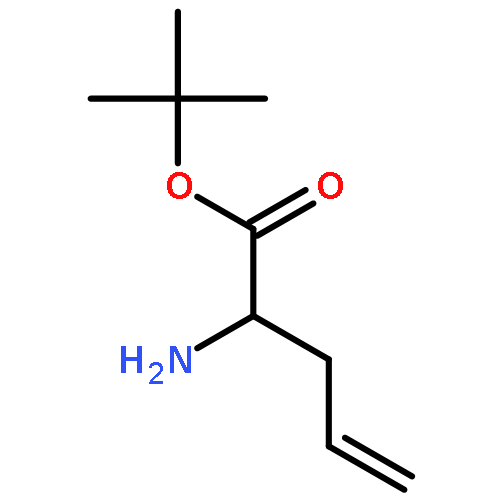
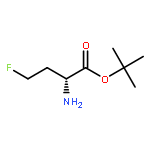
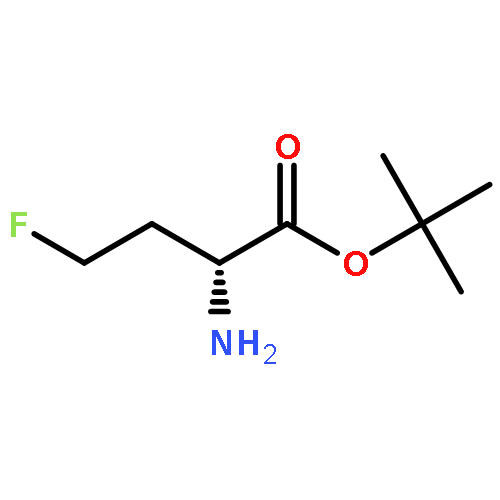






![1,3-Propanediol,2-amino-2-[2-(4-octylphenyl)ethyl]-](http://img.cochemist.com/ccimg/162400/162359-55-9.png)
![1,3-Propanediol,2-amino-2-[2-(4-octylphenyl)ethyl]-](http://img.cochemist.com/ccimg/162400/162359-55-9_b.png)

![Phosphonic acid, [2-methyl-1-[(phenylmethyl)amino]propyl]-, diethylester](http://img.cochemist.com/ccimg/131500/131479-91-9.png)
![Phosphonic acid, [2-methyl-1-[(phenylmethyl)amino]propyl]-, diethylester](http://img.cochemist.com/ccimg/131500/131479-91-9_b.png)
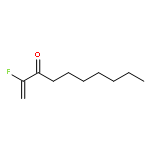
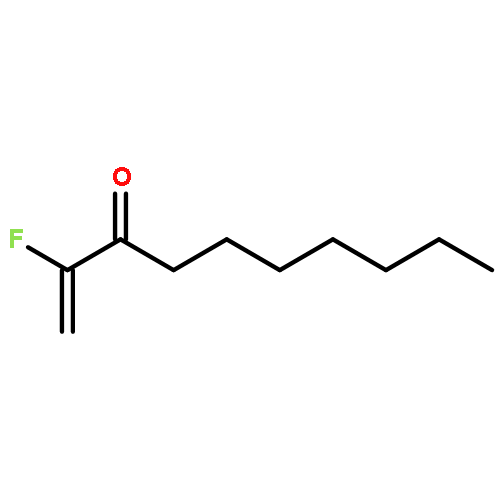
![Butanal, 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/87200/87184-81-4.png)
![Butanal, 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/87200/87184-81-4_b.png)
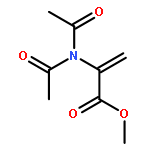
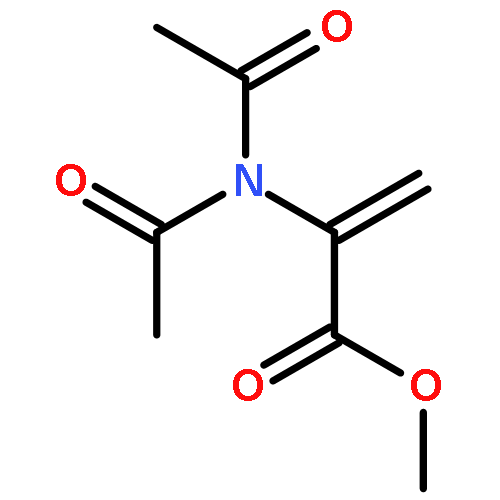
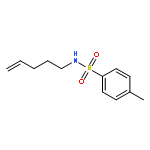
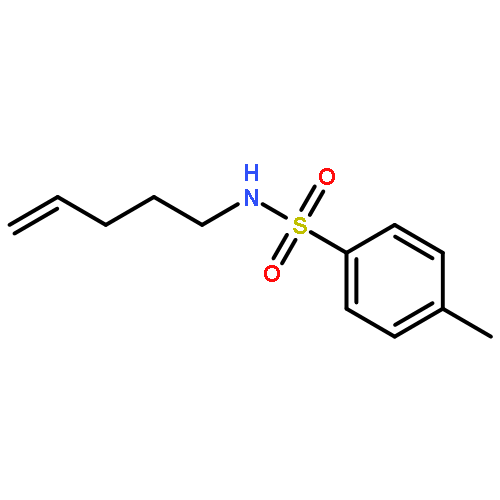
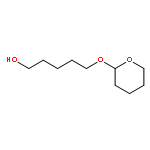

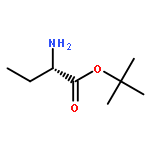
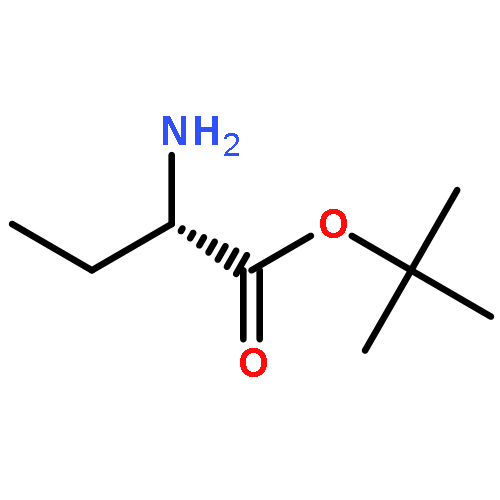
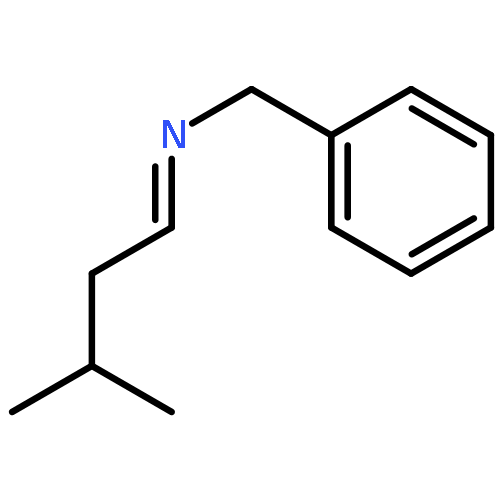
![Phosphonium, [(4-iodophenyl)methyl]triphenyl-, bromide](http://img.cochemist.com/ccimg/61200/61130-13-0.png)
![Phosphonium, [(4-iodophenyl)methyl]triphenyl-, bromide](http://img.cochemist.com/ccimg/61200/61130-13-0_b.png)
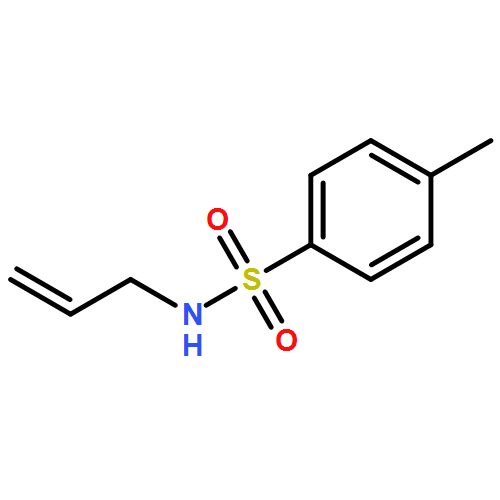
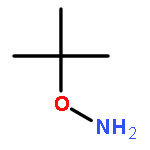
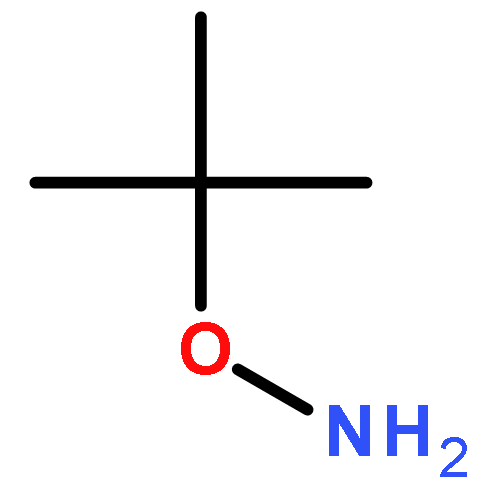
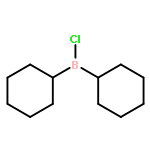
![1-Hexanol, 6-[(tetrahydro-2H-pyran-2-yl)oxy]-](http://img.cochemist.com/ccimg/28700/28659-22-5.png)
![1-Hexanol, 6-[(tetrahydro-2H-pyran-2-yl)oxy]-](http://img.cochemist.com/ccimg/28700/28659-22-5_b.png)
![BENZENE, 1-CHLORO-4-[(3-BROMOPROPYL)THIO]-](http://img.cochemist.com/ccimg/28000/27983-06-8.png)
![BENZENE, 1-CHLORO-4-[(3-BROMOPROPYL)THIO]-](http://img.cochemist.com/ccimg/28000/27983-06-8_b.png)
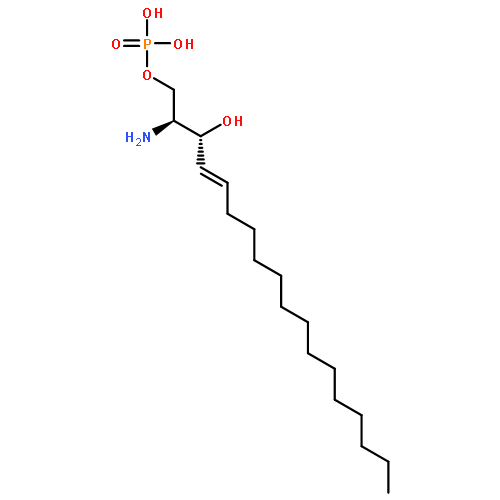
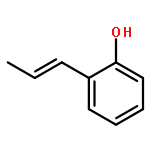
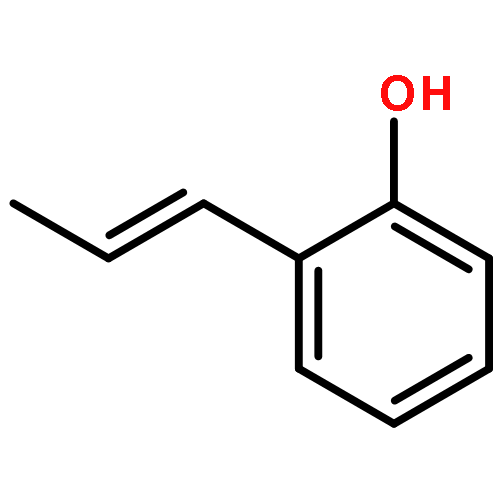
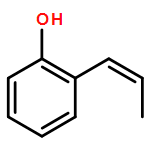

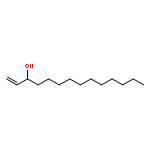
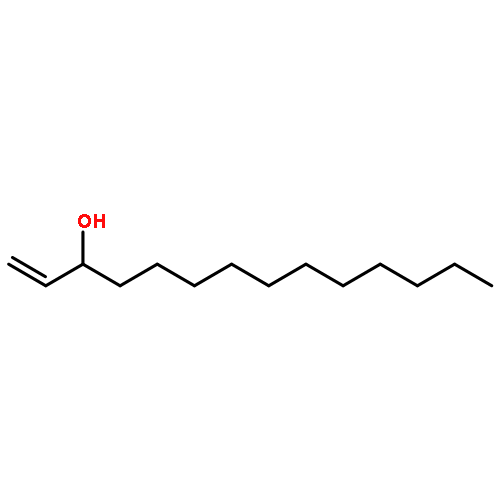
![Pentanal, 5-[(tetrahydro-2H-pyran-2-yl)oxy]-](http://img.cochemist.com/ccimg/14200/14194-86-6.png)
![Pentanal, 5-[(tetrahydro-2H-pyran-2-yl)oxy]-](http://img.cochemist.com/ccimg/14200/14194-86-6_b.png)

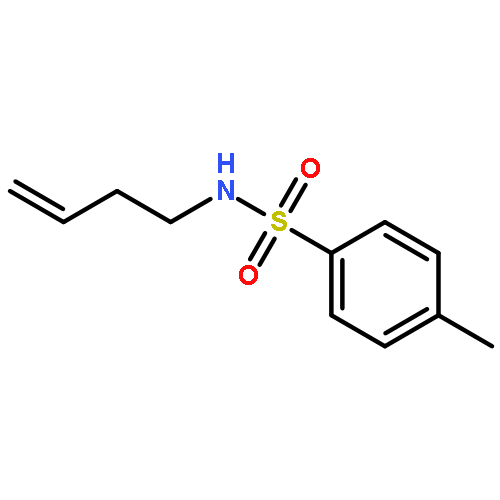
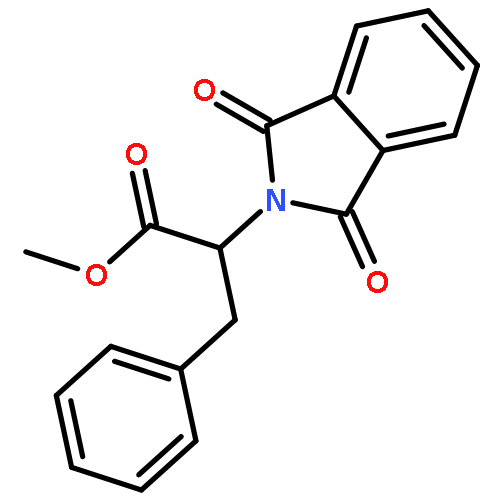
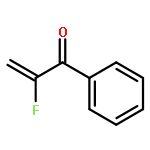
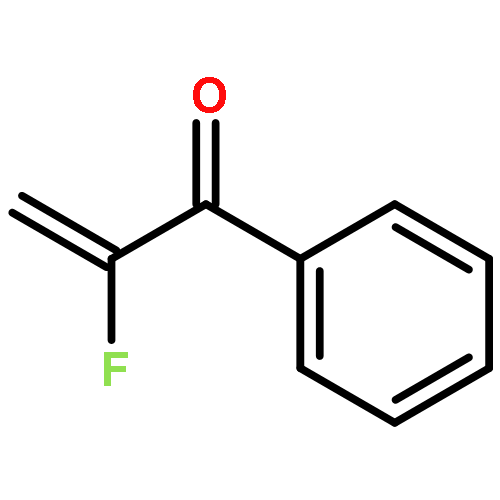
![Phosphonium,[(3-nitrophenyl)methyl]triphenyl-, bromide (1:1)](http://img.cochemist.com/ccimg/1600/1530-41-2.png)
![Phosphonium,[(3-nitrophenyl)methyl]triphenyl-, bromide (1:1)](http://img.cochemist.com/ccimg/1600/1530-41-2_b.png)