Co-reporter:Shoute Zhang, Weiquan Cai, Jiaguo Yu, Changchun Ji, Ning Zhao
Chemical Engineering Journal 2017 Volume 310, Part 1(Volume 310, Part 1) pp:
Publication Date(Web):15 February 2017
DOI:10.1016/j.cej.2016.10.114
•Supported MgO/γ-Al2O3 were prepared by a facile one-pot double hydrolysis method.•The MgO/γ-Al2O3 show enhanced CO2 adsorption capacities under simulated flue gas.•Mg/Al-0.2 shows a stable adsorption capacity after 11 cycles at 60 °C.•Bicarbonate, bidentate and monodentate carbonate are formed during CO2 adsorption.•Dynamic CO2 adsorption capacity increases with increasing the amount of basic sites.A series of supported MgO/γ-Al2O3 composites (denoted as Mg/Al-x) with enhanced adsorption performance towards CO2 were successfully synthesized via a facile one-pot cation-anion double hydrolysis approach. Their phase structures, morphologies, textural properties and basic properties were comparatively characterized by X-ray diffraction (XRD), scanning electron microscopy (SEM), transmission electron microscopy (TEM), selected area electron diffraction (SAED), energy dispersive X-ray spectroscopy (EDX), N2 adsorption-desorption, in situ diffuse reflection infrared Fourier transform spectroscopy (DRIFTS) and CO2 temperature programmed desorption (CO2-TPD). Three kinds of carbonates including bicarbonate, bidentate and monodentate carbonate are formed during the adsorption process, indicating a synergistic adsorption mechanism for CO2. For the dynamic CO2 adsorption without water vapor, the adsorption capacity decreases in the order of Mg/Al-0.2 > Mg/Al-0.3 > Mg/Al-0.1 > Mg/Al-0, which coincides with the trend of the amount of basic sites; especially, Mg/Al-0.2 with the Mg/Al molar ratio of 0.2 shows a stable adsorption capacity of 1.60 mmol/g without loss after 11 cycles at 60 °C. Furthermore, water vapor shows a positive effect on their adsorption, and Mg/Al-0.2 reaches an adsorption capacity of 2.10 mmol/g for the simulated flue gas with 10 vol% water vapor at 60 °C. Its relatively high adsorption capacity, excellent cyclic adsorption/desorption stability and facile preparation process make it an attractive candidate for CO2 capture with flue gas conditions.A series of supported MgO/γ-Al2O3 composites with enhanced CO2 adsorption capacity were successfully synthesized from Al(NO3)3·9H2O and Mg(NO3)2·6H2O using NaAlO2 as both precipitant and aluminium source simultaneously, and Pluronic P123 as structure directing agent via a facile one-pot cation-anion double hydrolysis approach. Among them, Mg/Al-0.2 with the Mg/Al molar ratio of 0.2 shows a stable adsorption capacity of 1.60 mmol/g without loss after 11 cycles under dynamic adsorption at 60 °C without water vapor, and further reaches an adsorption capacity of 2.10 mmol/g for the simulated flue gas with 10 vol% water vapor at 60 °C. Its relatively high adsorption capacity, good resistance to water vapor and excellent cyclic adsorption/desorption stability as well as its facile preparation process from cheap raw materials make it attractive candidate for CO2 capture under flue gas conditions.Download full-size image
Co-reporter:Xin Jin;Zhijun Cai
RSC Advances (2011-Present) 2017 vol. 7(Issue 84) pp:53076-53086
Publication Date(Web):2017/11/16
DOI:10.1039/C7RA10933D
Ordered mesoporous alumina (MA) was successfully modified with three amino organosilanes including 3-aminopropyltriethoxysilane (1N), N-(β-aminoethyl)-γ-aminopropylmethylbimethoxysilane (2N) and N-3-trimethoxysilylpropyldiethylenetriamine (3N) via a facile grafting method, and the as-prepared amino organosilane grafted MA–1N, MA–2N and MA–3N show enhanced adsorption performance towards Cr(VI) removal from wastewater. Their physicochemical properties and the MA before modification were comparatively characterized by FT-IR, TEM, XRD, N2 adsorption–desorption, CHNS elemental analysis, zeta potential measurements and XPS. Their adsorption performance was also comparatively studied along with the effect of contact time, adsorption isotherms, multi-metal ion adsorption, interference of co-existing anions and regeneration ability in batch experiments. It was found that their adsorption kinetics data were better fitted by the pseudo-second order model; adsorption isotherms were better described by the Langmuir isotherm for MA–1N, the Freundlich isotherm for MA–2N and the Temkin isotherm for MA–3N. Among them, MA–2N shows the maximum adsorption capacity of 137.9 mg g−1 which is more than twice the 59.4 mg g−1 of MA. The residual concentration of Cr(VI) with a concentration of 50 mg L−1 when treated with MA–2N meets the emission standard of the World Health Organization (WHO). Moreover, MA–2N shows better selectivity toward Cr(VI), and can reduce relatively more Cr(VI) to low toxicity Cr(III). More results were found that Cr(VI) is adsorbed on the surface through a monodentate ligand or bidentate ligand, and then Cr(VI) is reduced to Cr(III) by an adjacent electron donor, after which Cr(III) is co-precipitated with the adsorbents. All the amine-grafted samples show good reusability for 5 cycles. These results indicated that the amino organosilane grafted ordered MA with high adsorption capacity, good selectivity and favourable reusability is a promising candidate for Cr(VI) removal, in combination with its low cost and energy saving preparation process.
Co-reporter:Xuanjun Wu;Lei Li;Tiange Fang;YeTong Wang;Zhonghua Xiang
Physical Chemistry Chemical Physics 2017 vol. 19(Issue 13) pp:9261-9269
Publication Date(Web):2017/03/29
DOI:10.1039/C7CP01230F
By inserting an acetylene bond into the organic linkers of porous materials, hydrogen storage can be significantly enhanced; however, the mechanism of this enhancement remains elusive. Herein, we developed a new diamond-like carbon allotrope (referred as diamond-like diacetylene a.k.a. DDA) with medium pores constructed by inserting –CC–CC– ligands into the –C–C– bonds of diamond. The structural, mechanical, and electrical properties, as well as hydrogen storage capacities were investigated for this novel material using density functional theory and Monte Carlo simulations. The optimized geometry of DDA shows a high surface area and free pore volume of ca. 5498.76 m2 g−1 and 2.0486 m3 g−1, respectively. DDA also exhibits structural stability and special electronic properties. Interestingly, DDA exhibits exceptional gravimetric hydrogen storage capacity as well as volumetric one. The excess gravimetric and volumetric H2 uptakes at 77 K and 2.0 MPa hit a maximum of 14.12 wt% and 603.35 cm3 (STP) cm−3, respectively, which substantially exceeds those previously reported for MOF or PAF materials. Even at 243 K and 12 MPa, the total gravimetric H2 uptake of DDA reaches 5.38 wt%. To the best of our knowledge, DDA is one of porous materials with the maximum physical hydrogen uptake. It is also one of the few materials that can be close to meeting hydrogen storage target of the US department of energy at room temperature. Significantly, DDA shows the deliverable hydrogen storage capacity up to 5.28 wt% at room temperature. Through analyzing the effect of the acetylene position in the DLCAs on their hydrogen storage capacities, we found that the high hydrogen adsorption performance of DDA is mainly attributed to its high surface area, large number of adsorption sites, and appropriate binding energy. In summary, the newly developed DDA is a promising candidate for hydrogen storage and provides a new possibility for synthesizing high-performance adsorbents.
Co-reporter:Juan Fang, Debesh Devadutta Mishra, Weiquan Cai, Guolong Tan
Materials Science in Semiconductor Processing 2017 Volume 68(Volume 68) pp:
Publication Date(Web):1 September 2017
DOI:10.1016/j.mssp.2017.06.015
The present study explains the preparation of PbSnS3 nanocrystals using mechanical alloying as the processing technique and elemental powders as the starting material. The elemental powders of Pb, Sn and sublimed sulphur (S) were mechanically alloyed for 40 h. Phases evolved during mechanical alloying are explored by X-ray diffraction (XRD). The morphology and microstructural features have been investigated using High-resolution Transmission Electron Microscopy (HRTEM). UV–Vis–NIR spectroscopy has been used to measure the optical absorption characteristics. The mechanically alloyed powders show particle sizes in the range of 3–12 nm. The absorption edge extends from 503 nm to the visible region to 1360 nm near infrared region with a sudden change in slope at 860 nm, indicating the feature of indirect band gap semiconductors. The intersection point with the x-axis of extrapolating (αE)2 line as a function of E gives a direct band gap of 1.33 eV and an indirect gap of 0.59 eV.
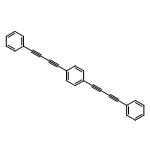
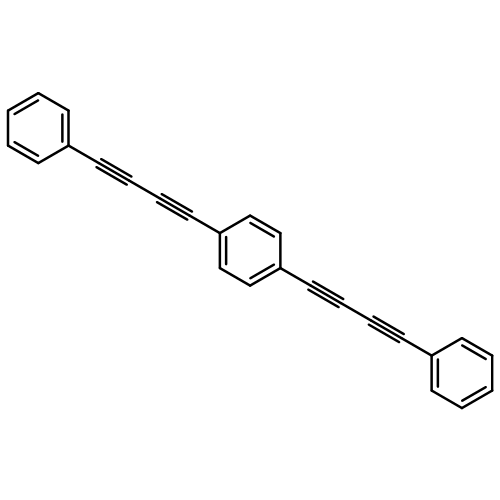
Co-reporter:Xuanjun Wu, Rui Wang, Hongjun Yang, Wenxuan Wang, Weiquan Cai and Qingzhong Li
Journal of Materials Chemistry A 2015 vol. 3(Issue 20) pp:10724-10729
Publication Date(Web):21 Apr 2015
DOI:10.1039/C5TA01290B
We proposed four novel porous aromatic frameworks (termed as PAF-322, PAF-324, PAF-332 and PAF-334) with low density and high free volume ratio, which were constructed by inserting a long and slim organic linker such as diphenylacetylene (DPA), 1,4-diphenyl-butadiyne (DPB), 1,4-bis (phenylethynyl) benzene (BPEB) or 1,4-bis (phenylbutadiynyl) benzene (BPBB) into each C–C bond of diamond. Then the hydrogen uptakes in these porous materials were predicted using grand canonical Monte Carlo (GCMC) simulations based on the force field derived from the first-principles calculations. The results show that these materials are the most promising candidates for hydrogen storage. Among the four novel PAFs, PAF-334 possesses the highest gravimetric hydrogen storage properties, which are a total gravimetric hydrogen uptake of 63.96 wt% at 77 K and 100 bar, and a excess gravimetric hydrogen uptake of 10.69 wt% at 77 K and 20 bar. In addition, the total gravimetric hydrogen uptake of these PAFs even at 243 K and 120 bar entirely exceeds the U.S. Department of Energy's (DOE) 2015 gravimetric hydrogen storage target. In particular, the total gravimetric hydrogen uptake of PAF-334 at 298 K and 100 bar reaches 16.03 wt%, about three times the DOE target value.
Co-reporter:Weiquan Cai, Yuzhen Hu, Jiaguo Yu, Wenguang Wang, Jiabin Zhou and Mietek Jaroniec
RSC Advances 2015 vol. 5(Issue 10) pp:7066-7073
Publication Date(Web):17 Dec 2014
DOI:10.1039/C4RA11329B
Hierarchical γ-Al2O3 nanostructures with tuneable morphologies including irregular nanoflake assemblies, melon-like nanoflake assemblies, flower-like ellipsoids, hollow core/shell and hollow microspheres were successfully synthesized for the first time via a facile template-free hydrothermal method using aluminium sulfate, aluminium chloride and aluminium nitrate as aluminium sources, respectively, and thiourea as precipitating agent. Their phase structures, morphologies, textural and basic properties were characterized by X-ray diffraction (XRD), scanning electron microscopy (SEM), transmission electron microscopy (TEM), selected area electron diffraction (SAED), N2 adsorption–desorption and CO2 temperature programmed desorption (CO2-TPD). The results indicate that the thiourea, type of anion in the aluminium source and the molar ratio of thiourea to Al3+ play an essential role in the formation of the aforementioned hierarchical γ-Al2O3. A growth mechanism of chemically induced self-transformation followed by cooperative self-assembly to form hierarchical nanostructures was proposed. In contrast, the γ-Al2O3 hollow core/shell microspheres with average pore size of 14.3 nm obtained from aluminium sulfate show the highest adsorption capacity of 28 mg g−1 towards phenol at 25 °C. However, the hierarchical γ-Al2O3 obtained from aluminium chloride and aluminium nitrate with smaller average pore size of 5.2 nm and 5.4 nm, respectively, is more effective for CO2 capture. This study provides new insights into the design and synthesis of hierarchical nanostructures for environmentally relevant applications.
Co-reporter:Xuanjun Wu, Jin Huang, Weiquan Cai and Mietek Jaroniec
RSC Advances 2014 vol. 4(Issue 32) pp:16503-16511
Publication Date(Web):18 Feb 2014
DOI:10.1039/C4RA00664J
A full set of flexible force field parameters for ZIF-8 is presented, based on the AMBER, UFF parameters and the partial charges computed by the density-derived electrostatic and chemical charge method (DDEC). The parameters for the 2-methyl imidazole (MeIM) ring are adopted from the AMBER force field, while the van der Waals (VDW) parameters for organic linkers and metal centers were determined by rescaling the UFF parameters as ε = 0.635εUFF and σ = 1.0σUFF to fit the CH4 adsorption isotherms obtained by Grand Canonical Monte Carlo (GCMC) simulations with the force field parameters to the experimental ones. The CH4 adsorption isotherms on four different structures of ZIF-8 at 298 K obtained by GCMC simulations are compared with the experimental data. The results show that the simulated CH4 adsorption isotherms on the ZIF-8 structure reported from the Cambridge Crystallographic Data Centre (CCDC) are closest to the ones on the ZIF-8 structure from the report of Moggach et al. To test our model, adsorption isotherms of CH4, H2, CO2 and N2 at different temperatures were computed using GCMC simulations, and the results were found to be in a good agreement with the experimental data. In the case of H2, the equilibrium configurations obtained by GCMC simulations were statistically analyzed with ad hoc code to get probability density distribution profiles. These profiles were transformed to visual slice images, which indicate that the preferential adsorption sites of H2 molecules in ZIF-8 are located close to the MeIM rings, where the host–guest VDW or electrostatic interactions are maximal, as revealed by the potential energy surfaces (PES). In addition, these force field parameters were confirmed to well reproduce the ZIF-8 structural properties including lattice constants, bond lengths and angles over a wide range of temperatures. The self-diffusivities at the specific loadings of adsorbed gases (CH4, H2 and CO2) in ZIF-8 were calculated by the mean squared displacement (MSD) method. It was found that our self-diffusivities of H2 are slightly higher than the ones in the literature, and our self-diffusivity of CO2 is as about three times as the one in the literature, due to the different partial charges and the effect of different force field parameters on framework shape and flexibility in our simulations.
Co-reporter:Jinrong Ge, Kejian Deng, Weiquan Cai, Jiaguo Yu, Xiaoqin Liu, Jiabin Zhou
Journal of Colloid and Interface Science 2013 Volume 401() pp:34-39
Publication Date(Web):1 July 2013
DOI:10.1016/j.jcis.2013.03.028
•Sphere- and flower-like γ-Al2O3 were hydrothermally prepared from sodium aluminate.•Morphologies of the γ-Al2O3 are dependent on the adopted structure-directing agents.•F127 mediated sphere-like γ-Al2O3 shows very fast adsorption kinetics toward Cr(VI).•Polyacrylic acid sodium mediated flower-like γ-Al2O3 shows good affinity toward CO2.Hierarchical flower-like and sphere-like mesoporous γ-Al2O3 microparticles were successfully prepared by a facile hydrothermal method followed by a calcination process using sodium aluminate as aluminum source, urea as precipitating agent, and Pluronic F127 (EO106PO70EO106), polyacrylic acid sodium (PAAS), and mixed F127–PAAS as structure-directing agents (SDAs), respectively. Effects of the SDAs on the phase structure, morphology, textural properties, surface alkaline, and the adsorption performance toward Cr(VI) and CO2 of the as-prepared samples were comparatively studied by X-ray diffraction (XRD), scanning electron microscope (SEM), transmission electron microscope (TEM), N2 adsorption–desorption, CO2 temperature programmed desorption (CO2-TPD), and UV–Vis spectrophotometric method. The results indicate that the sphere-like γ-Al2O3 obtained by using F127 as the SDA shows the best adsorption performance toward Cr(VI) with a high adsorption rate of 95% and adsorption capacity of 5.7 mg/g when the adsorption reaches equilibrium for 4 h at room temperature. However, the flower-like γ-Al2O3 obtained by using PAAS as the SDA has the biggest CO2 adsorption capacity of 1.04 mmol/g at room temperature. This work provides a simple and practical way to prepare potentially bifunctional γ-Al2O3 adsorbent for the removal of pollutants in water and air treatment from cheap sodium aluminate by using different SDAs.Graphical abstract
Co-reporter:Weiquan Cai, Shuanggui Chen, Jiaguo Yu, Yuzhen Hu, Chengxiong Dang, Shuhua Ma
Materials Chemistry and Physics 2013 Volume 138(Issue 1) pp:167-173
Publication Date(Web):15 February 2013
DOI:10.1016/j.matchemphys.2012.11.038
Three-dimensional hierarchical boehmite hollow microspheres with a very high yield at low cost were successfully synthesized via a one-pot template-free solvothermal route using aluminum chloride hexahydrate as precursor in a mixed ethanol–water solution with assistance of trisodium citrate. The as-synthesized products were characterized by X-ray powder diffraction (XRD), scanning electron microscopy (SEM), transmission electron microscopy (TEM), and nitrogen adsorption/desorption techniques. The results show that Cl− and addition amount of trisodium citrate have significant effect on the morphologies of the resultant products, and 6–8 mmol of trisodium citrate is optimal for the synthesis of boehmite hollow microspheres assembled from randomly interconnecting and aligned nanorods with solvothermal time no less than 15 h. A synergistic mediation mechanism of citrate ions and Cl− to form boehmite hollow spheres via self-assembly morphology evolution was proposed based on the experimental results. Interestingly, the typical boehmite hollow microspheres with a surface area of 102 m2 g−1, pore volume of 0.37 cm3 g−1, and the average pore size of 14.6 nm show superb adsorption properties for Congo red with maximum capacity of 114.7 mg g−1 which is higher than that of a commercial boehmite. This simple synthetic route is a very promising way for the design and synthesis of new functional hierarchical nanostructured materials with desired adsorptive properties.Graphical abstractBoehmite hollow microspheres with strong affinity toward organic pollutants were successfully synthesized via a one-pot template-free solvothermal route using AlCl3·6H2O as aluminum precursor in a mixed ethanol–water solution with assistance of trisodium citrate, and its adsorption performance toward Congo red is much higher than that of the commercial boehmite powder due to its unique hollow structure. Highlights► Boehmite hollow microspheres were solvothermally synthesized from AlCl3·6H2O. ► Diameter of the boehmite microspheres assembled from nanorods varies between 3 and 6 μm. ► The surface nanorods aligned in an ordered fashion show mean thick of 60 nm. ► Morphology changes of boehmite are dependent on the trisodium citrate concentration. ► The boehmite microspheres show good adsorption performance toward Congo red.
Co-reporter:Xuanjun Wu, Lei Li, Tiange Fang, YeTong Wang, Weiquan Cai and Zhonghua Xiang
Physical Chemistry Chemical Physics 2017 - vol. 19(Issue 13) pp:NaN9269-9269
Publication Date(Web):2017/03/06
DOI:10.1039/C7CP01230F
By inserting an acetylene bond into the organic linkers of porous materials, hydrogen storage can be significantly enhanced; however, the mechanism of this enhancement remains elusive. Herein, we developed a new diamond-like carbon allotrope (referred as diamond-like diacetylene a.k.a. DDA) with medium pores constructed by inserting –CC–CC– ligands into the –C–C– bonds of diamond. The structural, mechanical, and electrical properties, as well as hydrogen storage capacities were investigated for this novel material using density functional theory and Monte Carlo simulations. The optimized geometry of DDA shows a high surface area and free pore volume of ca. 5498.76 m2 g−1 and 2.0486 m3 g−1, respectively. DDA also exhibits structural stability and special electronic properties. Interestingly, DDA exhibits exceptional gravimetric hydrogen storage capacity as well as volumetric one. The excess gravimetric and volumetric H2 uptakes at 77 K and 2.0 MPa hit a maximum of 14.12 wt% and 603.35 cm3 (STP) cm−3, respectively, which substantially exceeds those previously reported for MOF or PAF materials. Even at 243 K and 12 MPa, the total gravimetric H2 uptake of DDA reaches 5.38 wt%. To the best of our knowledge, DDA is one of porous materials with the maximum physical hydrogen uptake. It is also one of the few materials that can be close to meeting hydrogen storage target of the US department of energy at room temperature. Significantly, DDA shows the deliverable hydrogen storage capacity up to 5.28 wt% at room temperature. Through analyzing the effect of the acetylene position in the DLCAs on their hydrogen storage capacities, we found that the high hydrogen adsorption performance of DDA is mainly attributed to its high surface area, large number of adsorption sites, and appropriate binding energy. In summary, the newly developed DDA is a promising candidate for hydrogen storage and provides a new possibility for synthesizing high-performance adsorbents.
Co-reporter:Xuanjun Wu, Rui Wang, Hongjun Yang, Wenxuan Wang, Weiquan Cai and Qingzhong Li
Journal of Materials Chemistry A 2015 - vol. 3(Issue 20) pp:NaN10729-10729
Publication Date(Web):2015/04/21
DOI:10.1039/C5TA01290B
We proposed four novel porous aromatic frameworks (termed as PAF-322, PAF-324, PAF-332 and PAF-334) with low density and high free volume ratio, which were constructed by inserting a long and slim organic linker such as diphenylacetylene (DPA), 1,4-diphenyl-butadiyne (DPB), 1,4-bis (phenylethynyl) benzene (BPEB) or 1,4-bis (phenylbutadiynyl) benzene (BPBB) into each C–C bond of diamond. Then the hydrogen uptakes in these porous materials were predicted using grand canonical Monte Carlo (GCMC) simulations based on the force field derived from the first-principles calculations. The results show that these materials are the most promising candidates for hydrogen storage. Among the four novel PAFs, PAF-334 possesses the highest gravimetric hydrogen storage properties, which are a total gravimetric hydrogen uptake of 63.96 wt% at 77 K and 100 bar, and a excess gravimetric hydrogen uptake of 10.69 wt% at 77 K and 20 bar. In addition, the total gravimetric hydrogen uptake of these PAFs even at 243 K and 120 bar entirely exceeds the U.S. Department of Energy's (DOE) 2015 gravimetric hydrogen storage target. In particular, the total gravimetric hydrogen uptake of PAF-334 at 298 K and 100 bar reaches 16.03 wt%, about three times the DOE target value.