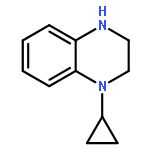
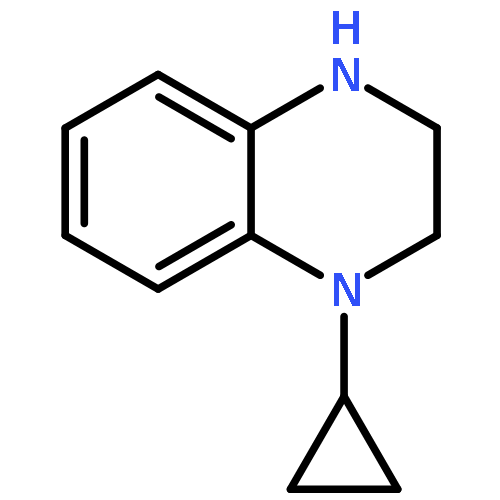
Co-reporter:Hongliang Duan; Mengmeng Ning; Qingan Zou; Yangliang Ye; Ying Feng; Lina Zhang; Ying Leng;Jianhua Shen
Journal of Medicinal Chemistry 2015 Volume 58(Issue 8) pp:3315-3328
Publication Date(Web):February 24, 2015
DOI:10.1021/jm500829b
Activation of TGR5 stimulates intestinal glucagon-like peptide-1 (GLP-1) release, but activation of the receptors in gallbladder and heart has been shown to cause severe on-target side effects. A series of low-absorbed TGR5 agonists was prepared by modifying compound 2 with polar functional groups to limit systemic exposure and specifically activate TGR5 in the intestine. Compound 15c, with a molecular weight of 1401, a PSA value of 223 Å2, and low permeability on Caco-2 cells, exhibited satisfactory potency both in vitro and in vivo. Low levels of 15c were detected in blood, bile, and gallbladder tissue, and gallbladder-related side effects were substantially decreased compared to the absorbed small-molecule TGR5 agonist 2.
Co-reporter:Junjie Zhu, Mengmeng Ning, Cen Guo, Lina Zhang, Guoyu Pan, Ying Leng, Jianhua Shen
European Journal of Medicinal Chemistry 2013 Volume 69() pp:55-68
Publication Date(Web):November 2013
DOI:10.1016/j.ejmech.2013.07.050
•A new class of TGR5 agonists based on a 4-phenyl pyridine scaffold was designed and synthesized.•Structure–activity relationships of this series were summarized.•Several compounds, like 15a, 18b and 18c, showed excellent potency in vitro.•15a displayed an unfavorable PK profile and was found to be ineffective during an OGTT in ICR mice at a dose of 50 mg/kg.TGR5, a GPCR, is involved in energy and glucose homeostasis, and as such, is a target for the treatment of diabetes, obesity and other metabolic syndromes. A new class of TGR5 agonists based on a 4-phenyl pyridine scaffold was designed, synthesized and evaluated in vitro and in vivo. Extensive structure–activity relationship studies are reported herein. The most potent compounds 15a, 18b and 18c showed comparable activity with the lead compound 2. 15a had the best potency in vitro but displayed an unfavorable pharmacokinetic profile and was found to be ineffective during an oral glucose tolerance test in imprinting control region mice at a dose of 50 mg/kg.A novel class of amides, sulfonamides and α-amino nitriles as potent TGR5 agonists was evaluated in vitro and in vivo.
Co-reporter:Junjie Zhu;Yangliang Ye;Mengmeng Ning;Attila Mándi;Ying Feng;Qingan Zou;Dr. Tibor Kurtán;Dr. Ying Leng;Dr. Jianhua Shen
ChemMedChem 2013 Volume 8( Issue 7) pp:1210-1223
Publication Date(Web):
DOI:10.1002/cmdc.201300144
Abstract
Given its role in the mediation of energy and glucose homeostasis, the G-protein-coupled bile acid receptor 1 (TGR5) is considered a potential target for the treatment of type 2 diabetes mellitus and other metabolic disorders. By thorough analysis of diverse structures of published TGR5 agonists, a hypothetical ligand-based pharmacophore model was built, and a new class of potent TGR5 agonists, based on the novel 3,4,5-trisubstituted 4,5-dihydro-1,2,4-oxadiazole core, was discovered by rational design. Three distinct synthetic methods for constructing 4,5-dihydro-1,2,4-oxadiazoles and extensive structure–activity relationship studies are reported herein. Compound (R)-54 n, the structure of which was determined by single-crystal X-ray diffraction and quantum chemical solid-state TDDFT-ECD calculations, showed the best potency, with an EC50 value of 1.4 nM toward hTGR5. Its favorable properties in vitro warrant further investigation.
Co-reporter:Hongliang Duan ; Mengmeng Ning ; Xiaoyan Chen ; Qingan Zou ; Liming Zhang ; Ying Feng ; Lina Zhang ; Ying Leng ;Jianhua Shen
Journal of Medicinal Chemistry 2012 Volume 55(Issue 23) pp:10475-10489
Publication Date(Web):November 13, 2012
DOI:10.1021/jm301071h
4-Phenoxynicotinamide and 4-phenoxypyrimidine-5-carboxamide derivatives as potent and orally efficacious TGR5 agonists are reported. Several 4-phenoxynicotinamide derivatives were found to activate human and mouse TGR5 (hTGR5 and mTGR5) with EC50 values in the low nanomolar range. Compound 23g, with an EC50 value of 0.72 nM on hTGR5 and an EC50 value of 6.2 nM on mTGR5, was selected for further in vivo efficacy studies. This compound exhibited a significant dose-dependent glucagon-like peptide-1 (GLP-1) secretion effect. A single oral dose of 23g (50 mg/kg) significantly reduced blood glucose levels in db/db mice and caused a 49% reduction in the area under the blood glucose curve (AUC)0–120 min following an oral glucose tolerance test (OGTT) in imprinting control region (ICR) mice. However, 23g stimulated gallbladder filling, which might result in side effects to the gallbladder.
Co-reporter:James D. Simo Mpetga, Yu Shen, Pierre Tane, Shi-Fei Li, Hong-Ping He, Hippolyte K. Wabo, Mathieu Tene, Ying Leng, and Xiao-Jiang Hao
Journal of Natural Products 2012 Volume 75(Issue 4) pp:599-604
Publication Date(Web):February 23, 2012
DOI:10.1021/np200831c
Five new triterpenoids, caloncobic acids A and B (1 and 2), caloncobalactones A and B (3 and 4), and glaucalactone (5), along with the known compounds 3β,21β-dihydroxy-30-nor-(D:A)-friedo-olean-20(29)-en-27-oic acid (6) and acetyltrichadenic acid B (7), were isolated from the leaves of Caloncoba glauca. The structures of 1–5 were elucidated using spectroscopic methods. Compounds 1–7 were evaluated for their inhibitory activities against two isozymes of 11β-hydroxysteroid dehydrogenase (11β-HSD1 and 11β-HSD2). Compounds 1 and 2 exhibited strong inhibitory activities against mouse (EC50 132 and 13 nM) and human (EC50 105 and 72 nM) 11β-HSD1.
Co-reporter:Na Ye, Xiangtao Xu, Fuying Li, Mengmeng Ning, Zhiqing Liu, Yuqing Cao, Ying Leng, Ao Zhang
Tetrahedron Letters 2012 Volume 53(Issue 35) pp:4738-4742
Publication Date(Web):29 August 2012
DOI:10.1016/j.tetlet.2012.06.111
RuCl3/NaIO4-initiated oxidation of oxiranemethanols was investigated, and two products, oxirane-2-carboxylic acid and (5′-oxotetrahydrofuran-3-yl)acetic acid, were obtained in variant ratios. The product structures were determined and a tentative mechanism involving oxidation-rearrangement-oxidation process was proposed. Glucokinase enzymatic assay revealed that oxiranecarboxamides 4a–c retained moderate GK activation potency with amide 4a showing an EC50 value of 584 nM and a high activation fold of 3.14. However, (5′-oxotetrahydrofuran-3-yl)acetamide 11a is inactive. This study not only provided an alternative protocol to access (5′-oxotetrahydrofuran-3-yl)acetic acid analogs, but also yielded nanomolar GA activators (esp. 4a) for further structure–activity relationship study.
Co-reporter:Liming Zhang, Junhua Chen, Mengmeng Ning, Qingan Zou, Ying Leng, Jianhua Shen
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 8) pp:2748-2752
Publication Date(Web):15 April 2012
DOI:10.1016/j.bmcl.2012.02.095
11β-Hydroxysteroid dehydrogenase type 1 (11β-HSD1) has attracted considerable attention as a potential target for the treatment of diabetes and metabolic syndrome. Herein we report the design, synthesis and efficacy evaluation of novel amide and urea 11β-HSD1 inhibitors. Structure–activity relationship studies led to the identification of 10c, which was efficacious in a diabetic ob/ob mouse model and reduced fasting and non-fasting blood glucose levels after ip dosing.Novel piperidine ureas were designed as potent 11β-HSD1 inhibitors. SAR studies led to the identification of 10c, which reduced fasting and non-fasting blood glucose levels after ip dosing in diabetic ob/ob mice.
Co-reporter:Weiwei Mao, Mengmeng Ning, Zhiqing Liu, Qingzhang Zhu, Ying Leng, Ao Zhang
Bioorganic & Medicinal Chemistry 2012 20(9) pp: 2982-2991
Publication Date(Web):
DOI:10.1016/j.bmc.2012.03.008
Co-reporter:Ying Feng;Su-ling Huang;Wei Dou;Song Zhang;Jun-hua Chen;Yu Shen;Jian-hua Shen
British Journal of Pharmacology 2010 Volume 161( Issue 1) pp:113-126
Publication Date(Web):
DOI:10.1111/j.1476-5381.2010.00826.x
BACKGROUND AND PURPOSE 11β-Hydroxysteroid dehydrogenase type 1 (11β-HSD1) is an attractive therapeutic target of type 2 diabetes and metabolic syndrome. Emodin, a natural product and active ingredient of various Chinese herbs, has been demonstrated to possess multiple biological activities. Here, we investigated the effects of emodin on 11β-HSD1 and its ability to ameliorate metabolic disorders in diet-induced obese (DIO) mice.
EXPERIMENTAL APPROACH Scintillation proximity assay was performed to evaluate inhibition of emodin against recombinant human and mouse 11β-HSDs. The ability of emodin to inhibit prednisone- or dexamethasone-induced insulin resistance was investigated in C57BL/6J mice and its effect on metabolic abnormalities was observed in DIO mice.
KEY RESULTS Emodin is a potent and selective 11β-HSD1 inhibitor with the IC50 of 186 and 86 nM for human and mouse 11β-HSD1, respectively. Single oral administration of emodin inhibited 11β-HSD1 activity of liver and fat significantly in mice. Emodin reversed prednisone-induced insulin resistance in mice, whereas it did not affect dexamethasone-induced insulin resistance, which confirmed its inhibitory effect on 11β-HSD1 in vivo. In DIO mice, oral administration of emodin improved insulin sensitivity and lipid metabolism, and lowered blood glucose and hepatic PEPCK, and glucose-6-phosphatase mRNA.
CONCLUSIONS AND IMPLICATIONS This study demonstrated a new role for emodin as a potent and selective inhibitor of 11β-HSD1 and its beneficial effects on metabolic disorders in DIO mice. This highlights the potential value of analogues of emodin as a new class of compounds for the treatment of metabolic syndrome or type 2 diabetes.
Co-reporter:Fuying Li, Qingzhang Zhu, Yi Zhang, Ying Feng, Ying Leng, Ao Zhang
Bioorganic & Medicinal Chemistry 2010 Volume 18(Issue 11) pp:3875-3884
Publication Date(Web):1 June 2010
DOI:10.1016/j.bmc.2010.04.038
A series of N-thiazole substituted arylacetamides were designed on the basis of metabolic mechanism of the aminothiazole fragment as glucokinase (GK) activators for the treatment of type 2 diabetes. Instead of introducing a substituent to block the metabolic sensitive C-5 position on the thiazole core directly, a wide variety of C-4 or both C-4 and C-5 substitutions were explored. Compound R-9k bearing an iso-propyl group as the C-4 substituent was found possessing the highest GK activation potency with an EC50 of 0.026 μM. This compound significantly increased both glucose uptake and glycogen synthesis in rat primary cultured hepatocytes. Moreover, single oral administration of compound R-9k exerted significant reduction of blood glucose levels in both ICR and ob/ob mice. These promising results indicated that compound R-9k is a potent orally active GK activator, and is warranted for further investigation as a new anti-diabetic treatment.A series of N-thiazole substituted arylacetamides were designed as metabolic stable glucokinase (GK) activators for the treatment of type 2 diabetes. Compound R-9k, with an EC50 of 0.026 μM significantly increased both glucose uptake and glycogen synthesis in rat primary cultured hepatocytes. Single oral administration of compound R-9k exerted significant reduction of blood glucose levels in both ICR and ob/ob mice.
Co-reporter:Ling Zhang, Yu Shen, Fei Wang, Ying Leng, Ji-Kai Liu
Phytochemistry 2010 Volume 71(Issue 1) pp:100-103
Publication Date(Web):January 2010
DOI:10.1016/j.phytochem.2009.09.020
Rare merosesquiterpenoids, craterellins A–C (1–3), were isolated from cultures of basidiomycete Craterellus odoratus together with the previously known massarinolin C (4). Structures of 1–3 were elucidated on the basis of extensive spectroscopic analysis. Compounds 1–3 possess a rare, epoxymethylenecyclohexanetriol-bicyclofarnesane sesquiterpene hybrid skeleton. Compounds 1–4 were evaluated for their inhibitory activities against two isozymes of 11β-hydroxysteroid dehydrogenases (11β-HSD1 and 11β-HSD2).Merosesquiterpenids, craterellins A–C (1–3), were isolated from cultures of the basidiomycete Craterellus odoratus together with the previously known massarinolin C (4). Compounds 1–3 possess a rare, epoxymethylenecyclohexanetriol-bicyclofarnesane sesquiterpene hybrid skeleton. Compounds 1–4 were evaluated for their inhibitory activities against 11β-hydroxysteroid dehydrogenases.
Co-reporter:Huaiyu Yang, Yu Shen, Junhua Chen, Qunfeng Jiang, Ying Leng, Jianhua Shen
European Journal of Medicinal Chemistry 2009 Volume 44(Issue 3) pp:1167-1171
Publication Date(Web):March 2009
DOI:10.1016/j.ejmech.2008.06.005
Structure-based pharmacophore models were built by using LigandScout and used for virtual screening of the SPECS database to identify new potential 11β-HSD1 inhibitors. As a refinement of the results obtained from virtual 3D pharmacophore screening, the best fitting virtual hits were subjected to docking study. The resulting compounds were tested in an enzyme assay and revealed several compounds with novel scaffolds that show sub-micromolar activity and high selectivity for 11β-HSD1 against 11β-HSD2.
Co-reporter:Yang-liang Ye, Zhou Zhou, Han-jun Zou, Yu Shen, Ti-fei Xu, Jing Tang, Hua-zhong Yin, Min-li Chen, Ying Leng, Jian-hua Shen
Bioorganic & Medicinal Chemistry 2009 Volume 17(Issue 15) pp:5722-5732
Publication Date(Web):1 August 2009
DOI:10.1016/j.bmc.2009.05.082
PPARγ and 11β-HSD1 are attractive therapeutic targets for type 2 diabetes. However, PPARγ agonists induce adipogenesis, which causes the side effect of weight gain, whereas 11β-HSD1 inhibitors prevent adipogenesis and may be beneficial for the treatment of obesity in diabetic patients. For the first time, we designed, synthesized a series of α-aryloxy-α-methylhydrocinnamic acids as dual functional agents which activate PPARγ and inhibit 11β-HSD1 simultaneously. The compound 11e exhibited the most potent inhibitory activity compared to that of the lead compound 2, with PPARγ (EC50 = 6.76 μM) and 11β-HSD1 (IC50 = 0.76 μM) in vitro. Molecular modeling study for compound 11e was also presented. Compound 11e showed excellent efficacy for lowering glucose, triglycerides, body fat, in well established mice and rats models of diabetes and obesity and had a favorable ADME profile.For the first time, we designed, synthesized a series of α-aryloxy-α-methylhydrocinnamic acids as dual functional agents which activate PPARγ and inhibit 11β-HSD1 simultaneously. The compound 11e displayed excellent hypoglycemic and hypolipidemic effect, but weak adipogenic activity with the decreased body weight and fat mass. Our finding suggests the dual functional compounds with PPARγ agonistic and 11β-HSD1 inhibitory activity might be a novel direction for anti-diabetic drug design.
Co-reporter:Ling Zhang, Yu Shen, Hua-Jie Zhu, Fei Wang, Ying Leng and Ji-Kai Liu
The Journal of Antibiotics 2009 62(5) pp:239-242
Publication Date(Web):March 27, 2009
DOI:10.1038/ja.2009.17
Five new secondary metabolites derived from pentanol, namely catathelasmols A–E (1–5), were isolated from the fruiting bodies of the basidiomycete Catathelasma imperiale. Their structures were elucidated on the basis of spectroscopic analysis, and the absolute configurations were determined by computational chemistry. Compounds 3, 4 and 5 showed inhibitory activities against two isozymes of 11-hydroxysteroid dehydrogenases (11-HSD1 and 11-HSD2), with IC50 values of 28.7–62.3 g ml−1 (human 11-HSD1), 30.4–149.2 g ml−1 (mouse 11-HSD1), 5.1–177 g ml−1 (human 11-HSD2) and 32.3–129.1 g ml−1 (mouse 11-HSD2), which catalyze the interconversion of cortisol and cortisone.
Co-reporter:Huaiyu Yang, Wei Dou, Jing Lou, Ying Leng, Jianhua Shen
Bioorganic & Medicinal Chemistry Letters 2008 Volume 18(Issue 4) pp:1340-1345
Publication Date(Web):15 February 2008
DOI:10.1016/j.bmcl.2008.01.020
11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1) is a potential target for treatment of diabetes and metabolic syndrome. Docking and pharmacophore modeling have been used to discover novel inhibitors of 11β-HSD1. Several compounds, with large structural diversity and good potency against 11β-HSD1, have been found and their potency was determined by the enzyme assay. New scaffolds of 11β-HSD1 inhibitors are also reported.Docking and pharmacophore Modeling were used to discover novel inhibitors of 11β-HSD1. Several compounds with large structural diversity and good potency were found. New scaffolds are reported.

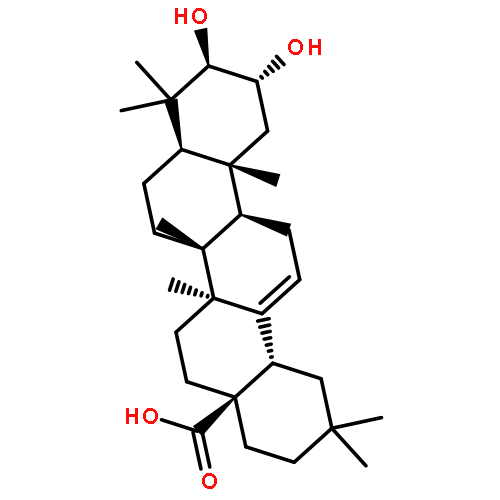
![(2S,4R)-4-[(3R,5S,6R,7R,8R,9S,10S,12S,13R,14S,17R)-6-ethyl-3,7,12-trihydroxy-10,13-dimethyl-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-17-yl]-2-methylpentanoic acid](http://img.cochemist.com/ccimg/1199800/1199796-29-6.png)
![(2S,4R)-4-[(3R,5S,6R,7R,8R,9S,10S,12S,13R,14S,17R)-6-ethyl-3,7,12-trihydroxy-10,13-dimethyl-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-17-yl]-2-methylpentanoic acid](http://img.cochemist.com/ccimg/1199800/1199796-29-6_b.png)
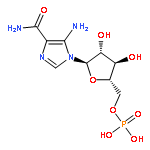
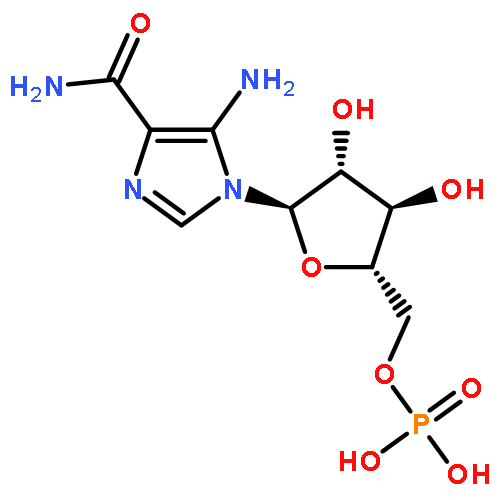
![2-Propanol, 1-[(1-methylethyl)amino]-3-(1-naphthalenyloxy)-](/data/chemimg/935900/525-66-6.png)
![2-Propanol, 1-[(1-methylethyl)amino]-3-(1-naphthalenyloxy)-](/data/chemimg/935900/525-66-6_b.png)






















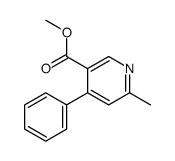
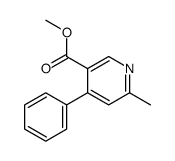
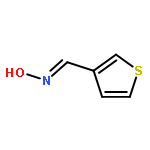
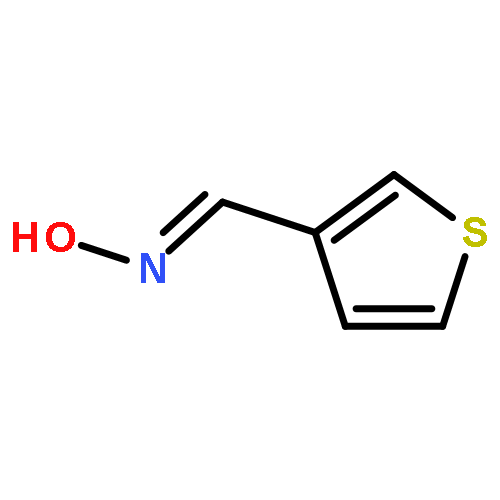
![Ethanamine, 2-[2-[2-(2-azidoethoxy)ethoxy]ethoxy]-](/data/chemimg/114300/134179-38-7.png)
![Ethanamine, 2-[2-[2-(2-azidoethoxy)ethoxy]ethoxy]-](/data/chemimg/114300/134179-38-7_b.png)
![Butanedioic acid, [(3-methoxyphenyl)methylene]-, 1-methyl ester](http://img.cochemist.com/ccimg/126700/126673-00-5.png)
![Butanedioic acid, [(3-methoxyphenyl)methylene]-, 1-methyl ester](http://img.cochemist.com/ccimg/126700/126673-00-5_b.png)




![4-METHOXY-4-OXO-3-[(3,4,5-TRIMETHOXYPHENYL)METHYL]BUTANOATE](/data/chemimg/394000/119138-93-1.png)
![4-METHOXY-4-OXO-3-[(3,4,5-TRIMETHOXYPHENYL)METHYL]BUTANOATE](/data/chemimg/394000/119138-93-1_b.png)
![4-[(4-METHOXYPHENYL)METHYL]OXOLAN-2-ONE](/data/chemimg/392700/118528-01-1.png)
![4-[(4-METHOXYPHENYL)METHYL]OXOLAN-2-ONE](/data/chemimg/392700/118528-01-1_b.png)
![Butanedioic acid, [(3-methoxyphenyl)methyl]-, 1-methyl ester](http://img.cochemist.com/ccimg/117900/117823-73-1.png)
![Butanedioic acid, [(3-methoxyphenyl)methyl]-, 1-methyl ester](http://img.cochemist.com/ccimg/117900/117823-73-1_b.png)




![Methyl [3,5-bis(trifluoromethyl)phenyl]acetate](http://img.cochemist.com/ccimg/95300/95299-16-4.png)
![Methyl [3,5-bis(trifluoromethyl)phenyl]acetate](http://img.cochemist.com/ccimg/95300/95299-16-4_b.png)
![2(5H)-FURANONE, DIHYDRO-4-[(3-METHOXYPHENYL)METHYL]-](http://img.cochemist.com/ccimg/86700/86653-16-9.png)
![2(5H)-FURANONE, DIHYDRO-4-[(3-METHOXYPHENYL)METHYL]-](http://img.cochemist.com/ccimg/86700/86653-16-9_b.png)
![Butanedioic acid, [(3,4,5-trimethoxyphenyl)methylene]-, 1-methyl ester](http://img.cochemist.com/ccimg/84800/84736-30-1.png)
![Butanedioic acid, [(3,4,5-trimethoxyphenyl)methylene]-, 1-methyl ester](http://img.cochemist.com/ccimg/84800/84736-30-1_b.png)
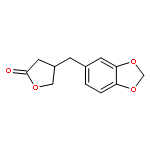
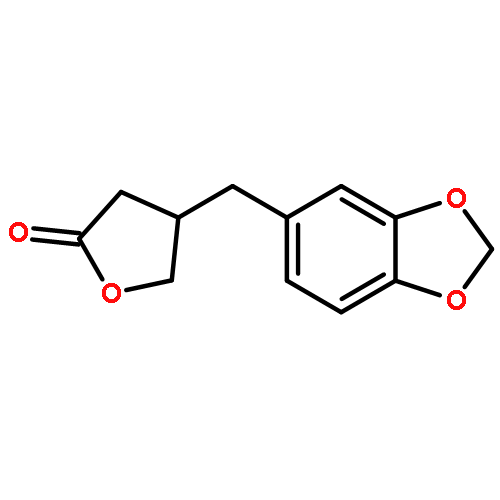
![Butanedioic acid, [(3,4-dimethoxyphenyl)methyl]-, 1-methyl ester](http://img.cochemist.com/ccimg/78700/78647-52-6.png)
![Butanedioic acid, [(3,4-dimethoxyphenyl)methyl]-, 1-methyl ester](http://img.cochemist.com/ccimg/78700/78647-52-6_b.png)

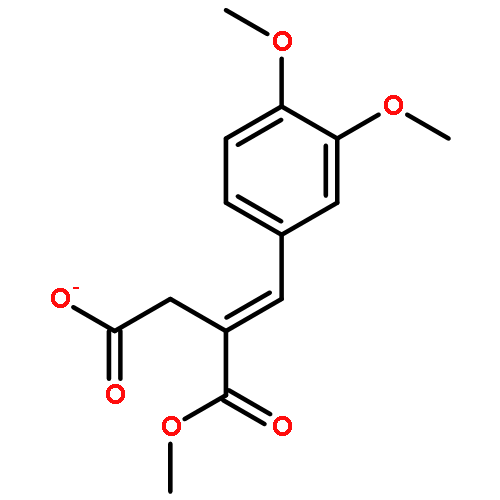


![2(3H)-Furanone, 4-[(3,4-dimethoxyphenyl)methyl]dihydro-](http://img.cochemist.com/ccimg/72500/72430-92-3.png)
![2(3H)-Furanone, 4-[(3,4-dimethoxyphenyl)methyl]dihydro-](http://img.cochemist.com/ccimg/72500/72430-92-3_b.png)
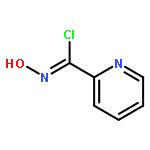
![2(3H)-FURANONE, DIHYDRO-4-[(3,4,5-TRIMETHOXYPHENYL)METHYL]-](http://img.cochemist.com/ccimg/65200/65171-10-0.png)
![2(3H)-FURANONE, DIHYDRO-4-[(3,4,5-TRIMETHOXYPHENYL)METHYL]-](http://img.cochemist.com/ccimg/65200/65171-10-0_b.png)
![Butanedioic acid, [(4-methoxyphenyl)methyl]-, 1-methyl ester](http://img.cochemist.com/ccimg/63200/63151-86-0.png)
![Butanedioic acid, [(4-methoxyphenyl)methyl]-, 1-methyl ester](http://img.cochemist.com/ccimg/63200/63151-86-0_b.png)
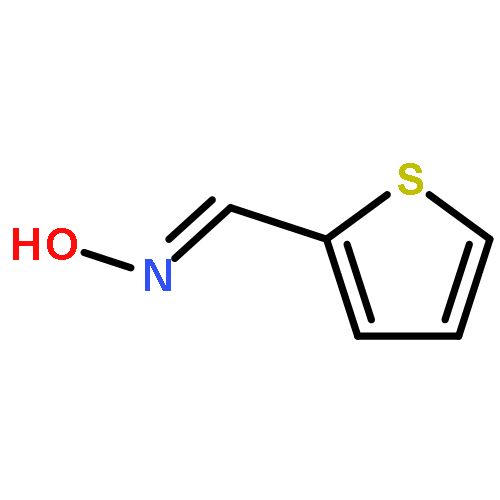
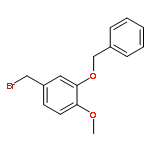
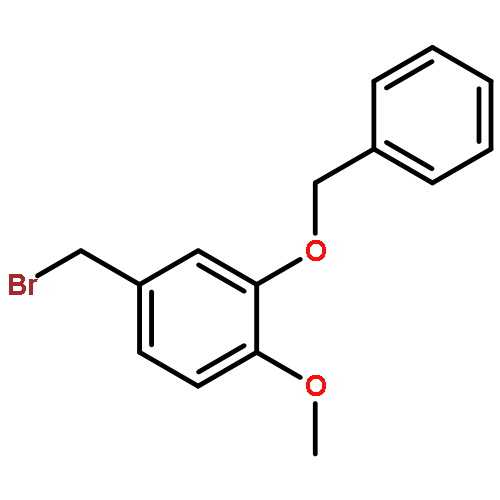
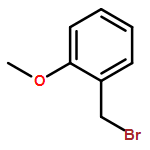
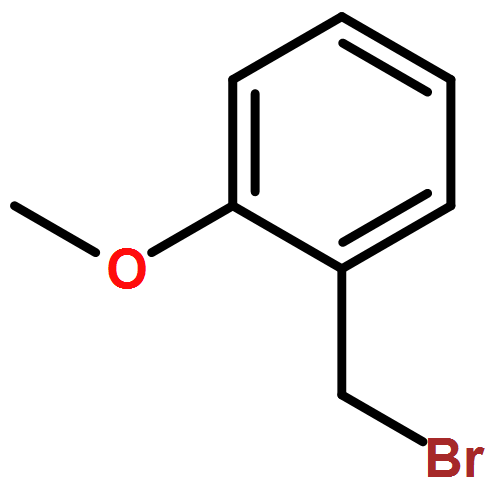
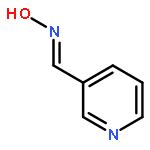
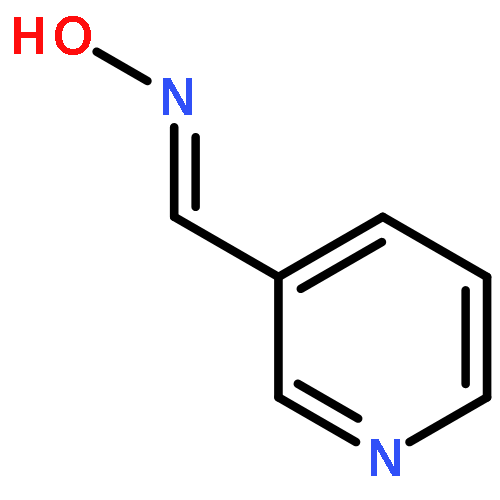
![Octadecanoic acid,1,1'-[1-(hydroxymethyl)-1,2-ethanediyl] ester](http://img.cochemist.com/ccimg/51100/51063-97-9.png)
![Octadecanoic acid,1,1'-[1-(hydroxymethyl)-1,2-ethanediyl] ester](http://img.cochemist.com/ccimg/51100/51063-97-9_b.png)
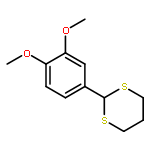


![Hexadecanoic acid,1,1'-[1-(hydroxymethyl)-1,2-ethanediyl] ester](http://img.cochemist.com/ccimg/40300/40290-32-2.png)
![Hexadecanoic acid,1,1'-[1-(hydroxymethyl)-1,2-ethanediyl] ester](http://img.cochemist.com/ccimg/40300/40290-32-2_b.png)





![Hexadecanoic acid,1,1'-[(1S)-1-(hydroxymethyl)-1,2-ethanediyl] ester](http://img.cochemist.com/ccimg/30400/30334-71-5.png)
![Hexadecanoic acid,1,1'-[(1S)-1-(hydroxymethyl)-1,2-ethanediyl] ester](http://img.cochemist.com/ccimg/30400/30334-71-5_b.png)

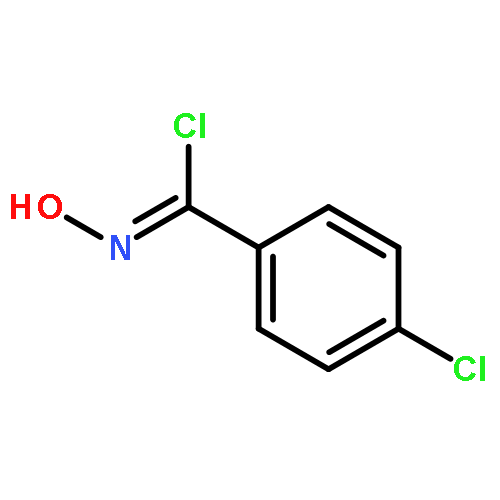
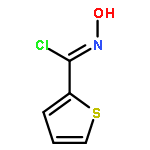
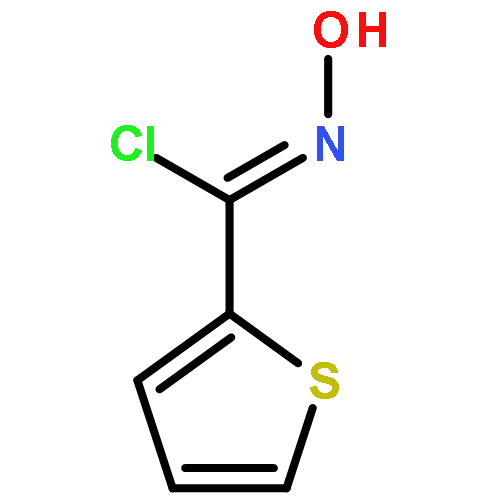
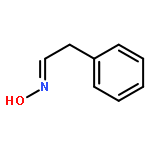
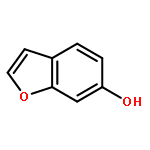
![Benzo[b]thiophen-5-ol](http://img.cochemist.com/ccimg/19400/19301-35-0.png)
![Benzo[b]thiophen-5-ol](http://img.cochemist.com/ccimg/19400/19301-35-0_b.png)
![Octadecanoic acid, 3-hydroxy-2-[(1-oxohexadecyl)oxy]propyl ester](http://img.cochemist.com/ccimg/14100/14015-55-5.png)
![Octadecanoic acid, 3-hydroxy-2-[(1-oxohexadecyl)oxy]propyl ester](http://img.cochemist.com/ccimg/14100/14015-55-5_b.png)
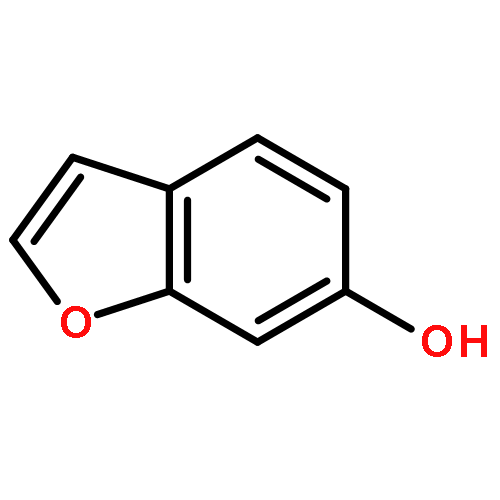




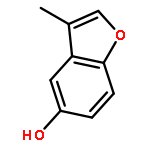
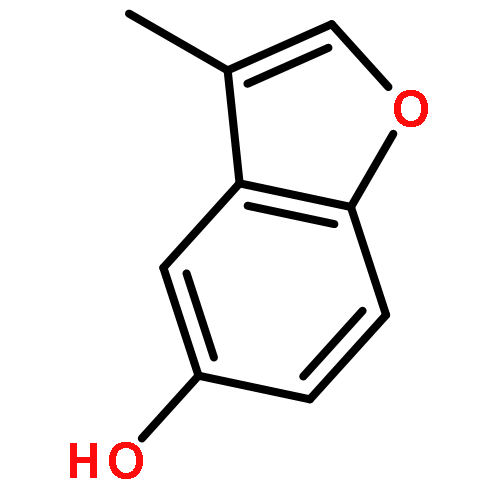


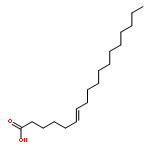
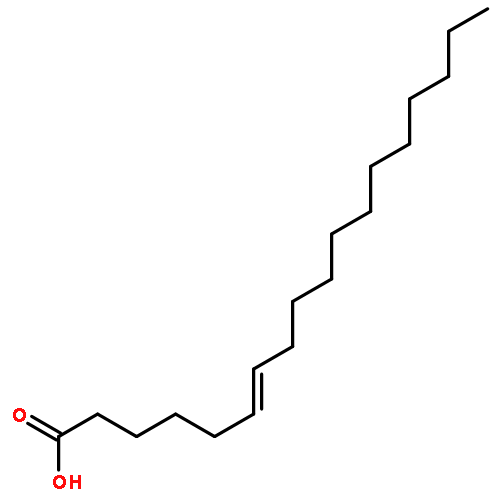
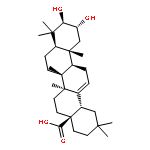
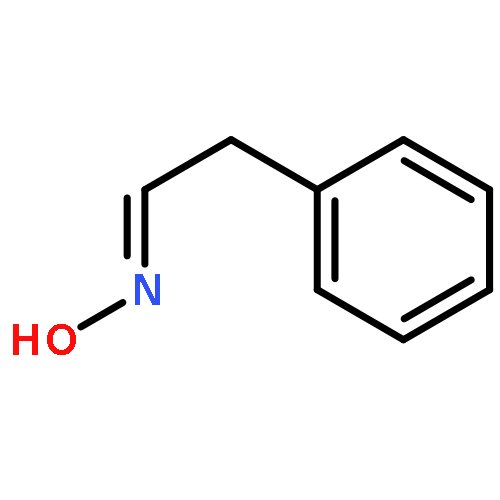
![9,12-Octadecadienoicacid (9Z,12Z)-, 1,1'-[1-(hydroxymethyl)-1,2-ethanediyl] ester](http://img.cochemist.com/ccimg/2500/2442-62-8.png)
![9,12-Octadecadienoicacid (9Z,12Z)-, 1,1'-[1-(hydroxymethyl)-1,2-ethanediyl] ester](http://img.cochemist.com/ccimg/2500/2442-62-8_b.png)


![[C(Z)]-2-Furancarboxaldehyde oxime](http://img.cochemist.com/ccimg/1500/1450-58-4.png)
![[C(Z)]-2-Furancarboxaldehyde oxime](http://img.cochemist.com/ccimg/1500/1450-58-4_b.png)



![5-TERT-BUTYL-2-[(4-FLUOROPHENYL)METHYL]PYRAZOLE-3-CARBOXYLIC ACID](http://img.cochemist.com/ccimg/700/620-03-1.png)
![5-TERT-BUTYL-2-[(4-FLUOROPHENYL)METHYL]PYRAZOLE-3-CARBOXYLIC ACID](http://img.cochemist.com/ccimg/700/620-03-1_b.png)