Co-reporter:Javier Iglesias-Fernández;Axel Kohlmeyer;Leonardo Darré;Robert K. Thomas;Hsin-Hui Shen
Langmuir October 13, 2015 Volume 31(Issue 40) pp:11097-11104
Publication Date(Web):2017-2-22
DOI:10.1021/acs.langmuir.5b02305
The lipopeptide surfactin produced by certain strains of Bacillus subtillis is a potent biosurfactant with high amphiphilicity and a strong tendency for self-aggregation. Surfactin possesses a number of valuable biological properties such as antiviral, antibacterial, antifungal, and hemolytic activities. Owing to these properties, in addition to the general advantages of biosurfactants over synthetic surfactants, surfactin has potential biotechnological and biomedical applications. Here, the aggregation properties of surfactin in solution together with its behavior at the water/air interface were studied using classical molecular dynamics simulations (MD) at three different pH values. Validation of the MD structural data was performed by comparing neutron reflectivity and volume fraction profiles computed from the simulations with their experimental counterparts. Analysis of the MD trajectories supported conclusions about the distribution, conformations, and interactions of surfactin in solution and at the water–air interface. Considering altogether, the work presented provides atomistic models for the rationalization of some of the structural and dynamic characteristics as well as the modes of action of surfactin at different pH values.
Co-reporter:Javier Iglesias-Fernandez, Peter J. Quinn, Richard J. Naftalin, Carmen Domene
Biophysical Journal 2017 Volume 112, Issue 6(Volume 112, Issue 6) pp:
Publication Date(Web):28 March 2017
DOI:10.1016/j.bpj.2017.01.030
Experimental evidence has shown a close correlation between the composition and physical state of the membrane bilayer and glucose transport activity via the glucose transporter GLUT1. Cooling alters the membrane lipids from the fluid to gel phase, and also causes a large decrease in the net glucose transport rate. The goal of this study is to investigate how the physical phase of the membrane alters glucose transporter structural dynamics using molecular-dynamics simulations. Simulations from an initial fluid to gel phase reduce the size of the cavities and tunnels traversing the protein and connecting the external regions of the transporter and the central binding site. These effects can be ascribed solely to membrane structural changes since in silico cooling of the membrane alone, while maintaining the higher protein temperature, shows protein structural and dynamic changes very similar to those observed with uniform cooling. These results demonstrate that the protein structure is sensitive to the membrane phase, and have implications for how transmembrane protein structures respond to their physical environment.
Co-reporter:Christian Jorgensen; Leonardo Darré; Victoria Oakes; Rubben Torella; David Pryde
Molecular Pharmaceutics 2016 Volume 13(Issue 7) pp:2263-2273
Publication Date(Web):May 12, 2016
DOI:10.1021/acs.molpharmaceut.5b00942
Potassium channels are of paramount physiological and pathological importance and therefore constitute significant drug targets. One of the keys to rationalize the way drugs modulate ion channels is to understand the ability of such small molecules to access their respective binding sites, from which they can exert an activating or inhibitory effect. Many computational studies have probed the energetics of ion permeation, and the mechanisms of voltage gating, but little is known about the role of fenestrations as possible mediators of drug entry in potassium channels. To explore the existence, structure, and conformational dynamics of transmembrane fenestrations accessible by drugs in potassium channels, molecular dynamics simulation trajectories were analyzed from three potassium channels: the open state voltage-gated channel Kv1.2, the G protein-gated inward rectifying channel GIRK2 (Kir3.2), and the human two-pore domain TWIK-1 (K2P1.1). The main results of this work were the identification of the sequence identity of four main lateral fenestrations of similar length and with bottleneck radius in the range of 0.9–2.4 Å for this set of potassium channels. It was found that the fenestrations in Kv1.2 and Kir3.2 remain closed to the passage of molecules larger than water. In contrast, in the TWIK-1 channel, both open and closed fenestrations are sampled throughout the simulation, with bottleneck radius shown to correlate with the random entry of lipid membrane molecules into the aperture of the fenestrations. Druggability scoring function analysis of the fenestration regions suggests that Kv and Kir channels studied are not druggable in practice due to steric constraining of the fenestration bottleneck. A high (>50%) fenestration sequence identity was found in each potassium channel subfamily studied, Kv1, Kir3, and K2P1. Finally, the reported fenestration sequence of TWIK-1 compared favorably with another channel, K2P channel TREK-2, reported to possess open fenestrations, suggesting that K2P channels could be druggable via fenestrations, for which we reported atomistic detail of the fenestration region, including the flexible residues M260 and L264 that interact with POPC membrane in a concerted fashion with the aperture and closure of the fenestrations.
Co-reporter:L. Darré, J. Iglesias-Fernandez, A. Kohlmeyer, H. Wacklin, and C. Domene
Journal of Chemical Theory and Computation 2015 Volume 11(Issue 10) pp:4875-4884
Publication Date(Web):September 17, 2015
DOI:10.1021/acs.jctc.5b00635
In combination with other spectroscopy, microscopy, and scattering techniques, neutron reflectivity is a powerful tool to characterize biological systems. Specular reflection of neutrons provides structural information at the nanometer and subnanometer length scales, probing the composition and organization of layered materials. Currently, analysis of neutron reflectivity data involves several simplifying assumptions about the structure of the sample under study, affecting the extraction and interpretation of information from the experimental data. Computer simulations can be used as a source of structural and dynamic data with atomic resolution. We present a novel tool to compare the structural properties determined by neutron reflectivity experiments with those obtained from molecular simulations. This tool allows benchmarking the ability of molecular dynamics simulations to reproduce experimental data, but it also promotes unbiased interpretation of experimentally determined quantities. Two application examples are presented to illustrate the capabilities of the new tool. The first example is the generation of reflectivity profiles for a 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC) lipid bilayer from molecular dynamics simulations using data from both atomistic and coarse-grained models, and comparison with experimentally measured data. The second example is the calculation of lipid volume changes with temperature and composition from all atoms simulations of single and mixed 1,2-di-palmitoyl-sn-glycero-3-phosphocholine (DOPC) and 1,2-dihexadecanoyl-sn-glycero-3-phosphocholine (DPPC) bilayers.
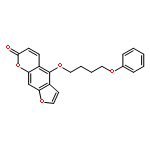
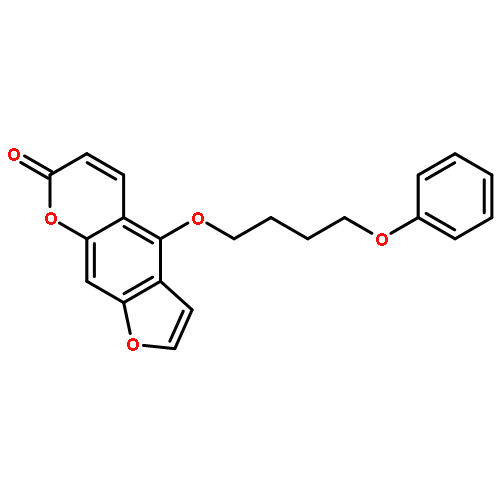
Co-reporter:Christian Jorgensen; Leonardo Darré; Kenno Vanommeslaeghe; Kiyoyuki Omoto; David Pryde
Molecular Pharmaceutics 2015 Volume 12(Issue 4) pp:1299-1307
Publication Date(Web):March 3, 2015
DOI:10.1021/acs.molpharmaceut.5b00023
Voltage-gated potassium channels of the Kv1 family play a crucial role in the generation and transmission of electrical signals in excitable cells affecting neuronal and cardiac activities. Small-molecule blockage of these channels has been proposed to occur via a cooperative mechanism involving two main blocking sites: the inner-pore site located below the selectivity filter, and a side-pocket cavity located between the pore and the voltage sensor. Using 0.5 μs molecular dynamics simulation trajectories complemented by docking calculations, the potential binding sites of the PAP-1 (5-(4-phenoxybutoxy)psoralen) blocker to the crystal structure of Kv1.2 channel have been studied. The presence of both mentioned blocking sites at Kv1.2 is confirmed, adding evidence in favor of a cooperative channel blockage mechanism. These observations provide insight into drug modulation that will guide further developments of Kv inhibitors.
Co-reporter:Leonardo Darré
Molecular Pharmaceutics 2015 Volume 12(Issue 12) pp:4454-4465
Publication Date(Web):October 26, 2015
DOI:10.1021/acs.molpharmaceut.5b00641
Transient receptor potential (TRP) ion channels constitute a notable family of cation channels involved in the ability of an organisms to detect noxious mechanical, thermal, and chemical stimuli that give rise to the perception of pain, taste, and changes in temperature. One of the most experimentally studied agonist of TRP channels is capsaicin, which is responsible for the burning sensation produced when chili pepper is in contact with organic tissues. Thus, understanding how this molecule interacts and regulates TRP channels is essential to high impact pharmacological applications, particularly those related to pain treatment. The recent publication of a three-dimensional structure of the vanilloid receptor 1 (TRPV1) in the absence and presence of capsaicin from single particle electron cryomicroscopy experiments provides the opportunity to explore these questions at the atomic level. In the present work, molecular docking and unbiased and biased molecular dynamics simulations were employed to generate a structural model of the capsaicin–channel complex. In addition, the standard free energy of binding was estimated using alchemical transformations coupled with conformational, translational, and orientational restraints on the ligand. Key binding modes consistent with previous experimental data are identified, and subtle but essential dynamical features of the binding site are characterized. These observations shed some light into how TRPV1 interacts with capsaicin, and may help to refine design parameters for new TRPV1 antagonists, and potentially guide further developments of TRP channel modulators.
Co-reporter:Simone Furini, Carmen Domene
Biochimica et Biophysica Acta (BBA) - Biomembranes (July 2016) Volume 1858(Issue 7) pp:
Publication Date(Web):1 July 2016
DOI:10.1016/j.bbamem.2016.02.015
Molecular dynamics simulations have played a fundamental role in numerous fields of science by providing insights into the structure and dynamics of complex systems at the atomistic level. However, exhaustive sampling by standard molecular dynamics is in most cases computationally prohibitive, and the time scales accessible remain significantly shorter than many biological processes of interest. In particular, in the study of ion channels, realistic models to describe permeation and gating require accounting for large numbers of particles and accurate interaction potentials, which severely limits the length of the simulations. To overcome such limitations, several advanced methods have been proposed among which is metadynamics. In this algorithm, an external bias potential to accelerate sampling along selected collective variables is introduced. This bias potential discourages visiting regions of the configurational space already explored. In addition, the bias potential provides an estimate of the free energy as a function of the collective variables chosen once the simulation has converged. In this review, recent contributions of metadynamics to the field of ion channels are discussed, including how metadynamics has been used to search for transition states, predict permeation pathways, treat conformational flexibility that underlies the coupling between gating and permeation, or compute free energy of permeation profiles. This article is part of a Special Issue entitled: Membrane Proteins edited by J.C. Gumbart and Sergei Noskov.
Co-reporter:Leonardo Darré, Simone Furini, Carmen Domene
Journal of Molecular Biology (30 January 2015) Volume 427(Issue 2) pp:537-549
Publication Date(Web):30 January 2015
DOI:10.1016/j.jmb.2014.11.016
•Ion conduction in TRPV1 was analyzed by molecular dynamics simulations.•Three main binding sites (SC, SF1 and SF2) define the permeation pathway.•Binding of Na+ and Ca2 + ions (but not K+) to SF2 links adjacent pore subunits.•The structure of the selectivity filter is sensible to the permeating ionic species.Transient receptor potential (TRP) ion channels constitute a large and diverse protein family, found in yeast and widespread in the animal kingdom. TRP channels work as sensors for a wide range of cellular and environmental signals. Understanding how these channels respond to physical and chemical stimuli has been hindered by the limited structural information available until now. The three-dimensional structure of the vanilloid receptor 1 (TRPV1) was recently determined by single particle electron cryo-microscopy, offering for the first time the opportunity to explore ionic conduction in TRP channels at atomic detail. In this study, we present molecular dynamics simulations of the open-activated pore domain of TRPV1 in the presence of three cationic species: Na+, Ca2 + and K+. The dynamics of these ions while interacting with the channel pore allowed us to rationalize their permeation mechanism in terms of a pathway involving three binding sites at the intracellular cavity, as well as the extracellular and intracellular entrance of the selectivity filter. Furthermore, conformational analysis of the pore in the presence of these ions reveals specific ion-mediated structural changes in the selectivity filter, which influences the permeability properties of the TRPV1 channel.Download high-res image (554KB)Download full-size image
Co-reporter:Victoria Oakes, Simone Furini, David Pryde, Carmen Domene
Biophysical Journal (23 August 2016) Volume 111(Issue 4) pp:
Publication Date(Web):23 August 2016
DOI:10.1016/j.bpj.2016.07.009
Potassium channels in the two-pore domain family (K2P) have various structural attributes that differ from those of other K+ channels, including a dimeric assembly constituted of nonidentical domains and an expansive extracellular cap. Crystallization of the prototypical K2P channel, TWIK-1, finally revealed the structure of these characteristics in atomic detail, allowing computational studies to be undertaken. In this study, we performed molecular-dynamics simulations for a cumulative time of ∼1 μs to discern the mechanism of ion transport throughout TWIK-1. We observed the free passage of ions beneath the extracellular cap and identified multiple high-occupancy sites in close proximity to charged residues on the protein surface. Despite the overall topological similarity of the x-ray structure of the selectivity filter to other K+ channels, the structure diverges significantly in molecular-dynamics simulations as a consequence of nonconserved residues in both pore domains contributing to the selectivity filter (T118 and L228). The behavior of such residues has been linked to channel inactivation and the phenomenon of dynamic selectivity, where TWIK-1 displays robust Na+ inward flux in response to subphysiological K+ concentrations.
Co-reporter:Christian Jorgensen, Simone Furini, Carmen Domene
Biophysical Journal (20 September 2016) Volume 111(Issue 6) pp:
Publication Date(Web):20 September 2016
DOI:10.1016/j.bpj.2016.08.009
Ion channels enable diffusion of ions down physiological electrochemical gradients. Modulation of ion permeation is crucial for the physiological functioning of cells, and misregulation of ion channels is linked to a myriad of channelopathies. The ion permeation mechanism in the transient receptor potential (TRP) ion channel family is currently not understood at an atomistic level. In this work, we employed a simulation strategy for ion permeation (molecular-dynamics simulations with bias-exchange metadynamics) to study and compare monovalent (Na+, K+) ion permeation in the open-activated TRP vanniloid-1 (TRPV1) ion channel. Using ∼3.6 μs of simulation trajectories, we obtained atomistic evidence for the nonselective nature of TRPV1. Our analysis shows that solvated monovalent ions permeate through the selectivity filter with comparable energetic barriers via a two-site mechanism. Finally, we confirmed that an intracellular binding site is located between the intracellular gate residues I679 and E684.
Co-reporter:Carmen Domene, Christian Jorgensen and Sumra Wajid Abbasi
Physical Chemistry Chemical Physics 2016 - vol. 18(Issue 36) pp:NaN24811-24811
Publication Date(Web):2016/08/17
DOI:10.1039/C6CP03403A
Collagen is the single most abundant protein in the extracellular matrix in the animal kingdom, with remarkable structural and functional diversity and regarded one of the most useful biomaterials. Etymologically, the term collagen comes from Greek kola ‘glue’ and gen ‘giving birth to’. Thus, it is not surprising that the various collagens and the structures they form all serve the same purpose, to help tissues withstand stretching. Among the functions the various collagens are involved in are cell adhesion and migration, tissue repair, scaffolding and morphogenesis. Thus knowledge about the structure and properties of collagen, how they change depending on the nature of the local environment as well as the nature and specificity of collagen interactions with its partners is central to discerning the role of collagen in medical applications such as imaging, drug delivery and tissue engineering, and in the design and construction of synthetic collagen-like materials for tools in biomaterial science and nanotechnology. The main focus of this perspective is to review the molecular and packing structures of collagen and the computer simulations work performed up to now to further highlight the significance of collagen.