Co-reporter:Khaleel I. Assaf, Mara Florea, Jens Antony, Niel M. Henriksen, Jian Yin, Andreas Hansen, Zheng-wang Qu, Rebecca Sure, Dieter Klapstein, Michael K. Gilson, Stefan Grimme, and Werner M. Nau
The Journal of Physical Chemistry B December 14, 2017 Volume 121(Issue 49) pp:11144-11144
Publication Date(Web):November 15, 2017
DOI:10.1021/acs.jpcb.7b09175
The host–guest complexation of hydrocarbons (22 guest molecules) with cucurbit[7]uril was investigated in aqueous solution using the indicator displacement strategy. The binding constants (103–109 M–1) increased with guest size, pointing to the hydrophobic effect and dispersion interactions as driving forces. The measured affinities provide unique benchmark data for the binding of neutral guest molecules. Consequently, a computational blind challenge, the HYDROPHOBE challenge, was conducted to allow a comparison with state-of-the-art computational methods for predicting host–guest affinity constants. In total, three quantum-chemical (QM) data sets and two explicit-solvent molecular dynamics (MD) submissions were received. When searching for sources of uncertainty in predicting the host–guest affinities, the experimentally known hydration energies of the investigated hydrocarbons were used to test the employed solvation models (explicit solvent for MD and COSMO-RS for QM). Good correlations were obtained for both solvation models, but a rather constant offset was observed for the COSMO data, by ca. +2 kcal mol–1, which was traced back to a required reference-state correction in the QM submissions (2.38 kcal mol–1). Introduction of the reference-state correction improved the predictive power of the QM methods, particularly for small hydrocarbons up to C5.
Co-reporter:Jakob Seibert, Christoph Bannwarth, and Stefan Grimme
Journal of the American Chemical Society August 30, 2017 Volume 139(Issue 34) pp:11682-11682
Publication Date(Web):August 11, 2017
DOI:10.1021/jacs.7b05833
A fully quantum mechanical (QM) treatment to calculate electronic absorption (UV–vis) and circular dichroism (CD) spectra of typical biomolecules with thousands of atoms is presented. With our highly efficient sTDA-xTB method, spectra averaged along structures from molecular dynamics (MD) simulations can be computed in a reasonable time frame on standard desktop computers. This way, nonequilibrium structure and conformational, as well as purely quantum mechanical effects like charge-transfer or exciton-coupling, are included. Different from other contemporary approaches, the entire system is treated quantum mechanically and neither fragmentation nor system-specific adjustment is necessary. Among the systems considered are a large DNA fragment, oligopeptides, and even entire proteins in an implicit solvent. We propose the method in tandem with experimental spectroscopy or X-ray studies for the elucidation of complex (bio)molecular structures including metallo-proteins like myoglobin.
Co-reporter:Christophe Werlé;Sebastian Dohm;Corinne Bailly;Lydia Karmazin;Louis Ricard;Nicolas Sieffert;Michel Pfeffer;Andreas Hansen;Jean-Pierre Djukic
Dalton Transactions 2017 vol. 46(Issue 25) pp:8125-8137
Publication Date(Web):2017/06/27
DOI:10.1039/C7DT00872D
Kinetically unstable heteroleptic trans-bispalladacycles were isolated by using hemichelation. Three structures of trans isomers and five of cis isomers were characterized by X-ray diffraction analysis. The ready trans-to-cis isomerization of such hemichelates that was monitored by variable temperature NMR experiments is facilitated dynamically because the Pd(II) center can preserve its square planar coordination in a rather low lying transition state, which was localized by methods of the density functional theory. This process is not achievable in the isomerization of conventional homoleptic trans-bispalladacycles since it involves the preliminary partial chelate decoordination and an unfavorable high-lying planar trigonal coordinated – or Y-shaped-Pd(II) transition state according to DFT investigations.
Co-reporter:Vilhjálmur Ásgeirsson;Christoph A. Bauer
Chemical Science (2010-Present) 2017 vol. 8(Issue 7) pp:4879-4895
Publication Date(Web):2017/06/26
DOI:10.1039/C7SC00601B
We introduce a fully stand-alone version of the Quantum Chemistry Electron Ionization Mass Spectra (QCEIMS) program [S. Grimme, Angew. Chem. Int. Ed., 2013, 52, 6306] allowing efficient simulations for molecules composed of elements with atomic numbers up to Z = 86. The recently developed extended tight-binding semi-empirical method GFN-xTB has been combined with QCEIMS, thereby eliminating dependencies on third-party electronic structure software. Furthermore, for reasonable calculations of ionization potentials, as required by the method, a second tight-binding variant, IPEA-xTB, is introduced here. This novel combination of methods allows the automatic, fast and reasonably accurate computation of electron ionization mass spectra for structurally different molecules across the periodic table. In order to validate and inspect the transferability of the method, we perform large-scale simulations for some representative organic, organometallic, and main-group inorganic systems. Theoretical spectra for 23 molecules are compared directly to experimental data taken from standard databases. For the first time, realistic quantum chemistry based EI-MS for organometallic systems like ferrocene or copper(II)acetylacetonate are presented. Compared to previously used semiempirical methods, GFN-xTB is faster, more robust, and yields overall higher quality spectra. The partially analysed theoretical reaction and fragmentation mechanisms are chemically reasonable and reveal in unprecedented detail the extreme complexity of high energy gas phase ion chemistry including complicated rearrangement reactions prior to dissociation.
Co-reporter:Dr. Yong-Qiang Zhang;Christina Poppel;Anastasia Panfilova;Fabian Bohle; Dr. Stefan Grimme; Dr. Andreas Gansäuer
Angewandte Chemie International Edition 2017 Volume 56(Issue 33) pp:9719-9722
Publication Date(Web):2017/08/07
DOI:10.1002/anie.201702882
AbstractDescribed herein is a novel concept for SN2 reactions at tertiary carbon centers in epoxides without activation of the leaving group. Quantum chemical calculations show why SN2 reactions at tertiary carbon centers are proceeding in these systems. The reaction allows flexible synthesis of 1,3-diol building blocks for natural product synthesis with excellent control of the relative and absolute configurations.
Co-reporter:Rebecca Sure;Andreas Hansen;Peter Schwerdtfeger
Physical Chemistry Chemical Physics 2017 vol. 19(Issue 22) pp:14296-14305
Publication Date(Web):2017/06/07
DOI:10.1039/C7CP00735C
We investigate all 1812 isomers of the fullerene C60 consistently with high-accuracy quantum chemistry methods. The isomers are optimized at the PBE-D3/def2-TZVP level of theory and their relative energies are obtained at the affordable and accurate hybrid-DFT level PW6B95-D3ATM/def2-QZVP. For the first time reliable values for the relative energy distribution among the 1812 isomers (maximum value of 549.1 kcal mol−1, average value of 189.8 ± 46.8 kcal mol−1, 1267 isomers in an energy window of 150–250 kcal mol−1) are given. The quality of the DFT energies is verified by comparison with highly accurate DLPNO-CCSD(T)/CBS* results for ten selected C60 isomers, and the DFT results are further used to benchmark several semiempirical methods. Our findings yield methodological insight into future multi-level modelling of larger carbon nano-structures. We correlate the best relative energies of the isomers to a number of topological indices, electronic properties, and geometrical measures in order to rationalize the isomeric stability aside from the common isolated pentagon rule minimizing the number of pentagon fusions. The information from the best qualitative measures can be condensed to the statement, that a small pentagon signature P1, a large volume, and a more spherical cage lead to a relatively stable isomer. In addition, the best semiempirical method was applied to all 31 924 isomers of C80.
Co-reporter: Dr. Stefan Grimme;M. Sc. Christoph Bannwarth;Dipl.-Chem. Sebastian Dohm;Dr. Andreas Hansen;M. Sc. Jana Pisarek;B. Sc. Philipp Pracht;M. Sc. Jakob Seibert; Dr. Frank Neese
Angewandte Chemie International Edition 2017 Volume 56(Issue 46) pp:14763-14769
Publication Date(Web):2017/11/13
DOI:10.1002/anie.201708266
AbstractWe present a composite procedure for the quantum-chemical computation of spin–spin-coupled 1H NMR spectra for general, flexible molecules in solution that is based on four main steps, namely conformer/rotamer ensemble (CRE) generation by the fast tight-binding method GFN-xTB and a newly developed search algorithm, computation of the relative free energies and NMR parameters, and solving the spin Hamiltonian. In this way the NMR-specific nuclear permutation problem is solved, and the correct spin symmetries are obtained. Energies, shielding constants, and spin–spin couplings are computed at state-of-the-art DFT levels with continuum solvation. A few (in)organic and transition-metal complexes are presented, and very good, unprecedented agreement between the theoretical and experimental spectra was achieved. The approach is routinely applicable to systems with up to 100–150 atoms and may open new avenues for the detailed (conformational) structure elucidation of, for example, natural products or drug molecules.
Co-reporter:Lars Goerigk;Andreas Hansen;Christoph Bauer;Stephan Ehrlich;Asim Najibi
Physical Chemistry Chemical Physics 2017 vol. 19(Issue 48) pp:32184-32215
Publication Date(Web):2017/12/13
DOI:10.1039/C7CP04913G
We present the GMTKN55 benchmark database for general main group thermochemistry, kinetics and noncovalent interactions. Compared to its popular predecessor GMTKN30 [Goerigk and Grimme J. Chem. Theory Comput., 2011, 7, 291], it allows assessment across a larger variety of chemical problems—with 13 new benchmark sets being presented for the first time—and it also provides reference values of significantly higher quality for most sets. GMTKN55 comprises 1505 relative energies based on 2462 single-point calculations and it is accessible to the user community via a dedicated website. Herein, we demonstrate the importance of better reference values, and we re-emphasise the need for London-dispersion corrections in density functional theory (DFT) treatments of thermochemical problems, including Minnesota methods. We assessed 217 variations of dispersion-corrected and -uncorrected density functional approximations, and carried out a detailed analysis of 83 of them to identify robust and reliable approaches. Double-hybrid functionals are the most reliable approaches for thermochemistry and noncovalent interactions, and they should be used whenever technically feasible. These are, in particular, DSD-BLYP-D3(BJ), DSD-PBEP86-D3(BJ), and B2GPPLYP-D3(BJ). The best hybrids are ωB97X-V, M052X-D3(0), and ωB97X-D3, but we also recommend PW6B95-D3(BJ) as the best conventional global hybrid. At the meta-generalised-gradient (meta-GGA) level, the SCAN-D3(BJ) method can be recommended. Other meta-GGAs are outperformed by the GGA functionals revPBE-D3(BJ), B97-D3(BJ), and OLYP-D3(BJ). We note that many popular methods, such as B3LYP, are not part of our recommendations. In fact, with our results we hope to inspire a change in the user community's perception of common DFT methods. We also encourage method developers to use GMTKN55 for cross-validation studies of new methodologies.
Co-reporter:Dr. Yong-Qiang Zhang;Christina Poppel;Anastasia Panfilova;Fabian Bohle; Dr. Stefan Grimme; Dr. Andreas Gansäuer
Angewandte Chemie 2017 Volume 129(Issue 33) pp:9851-9854
Publication Date(Web):2017/08/07
DOI:10.1002/ange.201702882
AbstractDescribed herein is a novel concept for SN2 reactions at tertiary carbon centers in epoxides without activation of the leaving group. Quantum chemical calculations show why SN2 reactions at tertiary carbon centers are proceeding in these systems. The reaction allows flexible synthesis of 1,3-diol building blocks for natural product synthesis with excellent control of the relative and absolute configurations.
Co-reporter:Stefan Grimme, Andreas Hansen, Jan Gerit Brandenburg, and Christoph Bannwarth
Chemical Reviews 2016 Volume 116(Issue 9) pp:5105
Publication Date(Web):April 14, 2016
DOI:10.1021/acs.chemrev.5b00533
Mean-field electronic structure methods like Hartree–Fock, semilocal density functional approximations, or semiempirical molecular orbital (MO) theories do not account for long-range electron correlation (London dispersion interaction). Inclusion of these effects is mandatory for realistic calculations on large or condensed chemical systems and for various intramolecular phenomena (thermochemistry). This Review describes the recent developments (including some historical aspects) of dispersion corrections with an emphasis on methods that can be employed routinely with reasonable accuracy in large-scale applications. The most prominent correction schemes are classified into three groups: (i) nonlocal, density-based functionals, (ii) semiclassical C6-based, and (iii) one-electron effective potentials. The properties as well as pros and cons of these methods are critically discussed, and typical examples and benchmarks on molecular complexes and crystals are provided. Although there are some areas for further improvement (robustness, many-body and short-range effects), the situation regarding the overall accuracy is clear. Various approaches yield long-range dispersion energies with a typical relative error of 5%. For many chemical problems, this accuracy is higher compared to that of the underlying mean-field method (i.e., a typical semilocal (hybrid) functional like B3LYP).
Co-reporter:Weichao Xue, Zheng-Wang Qu, Stefan Grimme, and Martin Oestreich
Journal of the American Chemical Society 2016 Volume 138(Issue 43) pp:14222-14225
Publication Date(Web):October 16, 2016
DOI:10.1021/jacs.6b09596
A copper-catalyzed C(sp3)–Si cross-coupling of aliphatic C(sp3)–I electrophiles using a Si–B reagent as the silicon pronucleophile is reported. The reaction involves an alkyl radical intermediate that also engages in 5-exo-trig ring closures onto pendant alkenes prior to the terminating C(sp3)–Si bond formation. Several Ueno–Stork-type precursors cyclized with excellent diastereocontrol in good yields. The base-mediated release of the silicon nucleophile and the copper-catalyzed radical process are analyzed by quantum-chemical calculations, leading to a full mechanistic picture.
Co-reporter:Rebecca Sure and Stefan Grimme
Chemical Communications 2016 vol. 52(Issue 64) pp:9893-9896
Publication Date(Web):07 Jul 2016
DOI:10.1039/C6CC03664C
Recently, Diederich et al. synthesized the first supramolecular capsule with a well-defined four-point halogen bonding interaction [Angew. Chem., Int. Ed., 2015, 54, 12339]. This interesting system comprising about 400 atoms represents a challenging test case for accurate quantum chemical methods. We investigate it with our new density functional based composite method for structures and noncovalent interactions (PBEh-3c) as well as our standard protocol for supramolecular thermochemistry and give predictions for chemical modifications to improve the binding strength.
Co-reporter:Stefan Grimme and Marc Steinmetz
Physical Chemistry Chemical Physics 2016 vol. 18(Issue 31) pp:20926-20937
Publication Date(Web):03 Dec 2015
DOI:10.1039/C5CP06600J
We present a revised form of a double hybrid density functional (DHDF) dubbed PWRB95. It contains semi-local Perdew–Wang exchange and Becke95 correlation with a fixed amount of 50% non-local Fock exchange. New features are that the robust random phase approximation (RPA) is used to calculate the non-local correlation part instead of a second-order perturbative treatment as in standard DHDF, and the non-self-consistent evaluation of the Fock exchange with KS-orbitals at the GGA level which leads to a significant reduction of the computational effort. To account for London dispersion effects we include the non-local VV10 dispersion functional. Only three empirical scaling parameters were adjusted. The PWRB95 results for extensive standard thermochemical benchmarks (GMTKN30 data base) are compared to those of well-known functionals from the classes of (meta-)GGAs, (meta-)hybrid functionals, and DHDFs, as well as to standard (direct) RPA. The new method is furthermore tested on prototype bond activations with (Ni/Pd)-based transition metal catalysts, and two difficult cases for DHDF, namely the isomerization reaction of the [Cu2(en)2O2]2+ complex and the singlet–triplet energy difference in highly unsaturated cyclacenes. The results show that PWRB95 is almost as accurate as standard DHDF for main-group thermochemistry but has a similar or better performance for non-covalent interactions, more difficult transition metal containing molecules and other electronically problematic cases. Because of its relatively weak basis set dependence, PWRB95 can be applied even in combination with AO basis sets of only triple-zeta quality which yields huge overall computational savings by a factor of about 40 compared to standard DHDF/‘quadruple-zeta’ calculations. Structure optimizations of small molecules with PWRB95 indicate an accurate description of bond distances superior to that provided by TPSS-D3, PBE0-D3, or other RPA type methods.
Co-reporter:Jan Gerit Brandenburg, Eike Caldeweyher and Stefan Grimme
Physical Chemistry Chemical Physics 2016 vol. 18(Issue 23) pp:15519-15523
Publication Date(Web):24 May 2016
DOI:10.1039/C6CP01697A
We extend the recently introduced PBEh-3c global hybrid density functional [S. Grimme et al., J. Chem. Phys., 2015, 143, 054107] by a screened Fock exchange variant based on the Henderson–Janesko–Scuseria exchange hole model. While the excellent performance of the global hybrid is maintained for small covalently bound molecules, its performance for computed condensed phase mass densities is further improved. Most importantly, a speed up of 30 to 50% can be achieved and especially for small orbital energy gap cases, the method is numerically much more robust. The latter point is important for many applications, e.g., for metal–organic frameworks, organic semiconductors, or protein structures. This enables an accurate density functional based electronic structure calculation of a full DNA helix structure on a single core desktop computer which is presented as an example in addition to comprehensive benchmark results.
Co-reporter:Christian Honacker, Zheng-Wang Qu, Jens Tannert, Marcus Layh, Alexander Hepp, Stefan Grimme and Werner Uhl
Dalton Transactions 2016 vol. 45(Issue 14) pp:6159-6174
Publication Date(Web):19 Nov 2015
DOI:10.1039/C5DT03918E
Treatment of alkynyl–arylchlorogermanes ArylnGe(Cl)(CC–tBu)3−n (n = 1, 2) with HMtBu2 (M = Al, Ga) yielded mixed Al or Ga alkenyl–alkynylchlorogermanes via hydrometallation reactions. Intramolecular interactions between the Lewis-basic Cl atoms and the Lewis-acidic Al or Ga atoms afforded MCGeCl heterocycles. The endocyclic M–Cl distances were significantly lengthened compared to the starting compounds and indicated Ge–Cl bond activation. Dual hydrometallation succeeded only with HGatBu2. One Ga atom of the product was involved in a Ga–Cl bond, while the second one had an interaction to a C–H bond of a phenyl group. In two cases treatment of chlorogermanes with two equivalents of HAltBu2 resulted in hydroalumination of one alkynyl group and formation of unprecedented Ge–H functionalized germanes, Aryl–Ge(H)(CC–tBu)[C(AltBu2)C(H)–tBu] (Aryl = mesityl, triisopropylphenyl). The Al atoms of these compounds interacted with the α-C atoms of the alkynyl groups. Ph(Cl)Ge(CC–tBu)[C(AltBu2}C(H)–tBu] reacted in an unusual Cl/tBu exchange to yield the tert-butylgermane Ph(tBu)Ge(CC–tBu)[C{Al(tBu)(Cl)}C(H)–tBu]. Quantum chemical calculations suggested the formation of a germyl cation as a transient intermediate.
Co-reporter:Christopher Schweez;Dr. Philip Shushkov;Dr. Stefan Grimme;Dr. Sigurd Höger
Angewandte Chemie 2016 Volume 128( Issue 10) pp:3389-3394
Publication Date(Web):
DOI:10.1002/ange.201509702
Abstract
Phenylacetylene-based [2]rotaxanes were synthesized by a covalent-template approach by aminolysis of the corresponding prerotaxanes. The wheel and the bulky stoppers are made of phenylene–ethynylene–butadiynylene macrocycles of the same size. The stoppers are large enough to enable the synthesis and purification of the rotaxane. However, the wheel unthreads from the axle at elevated temperatures. The deslipping kinetics and the activation parameters were determined. We described theoretically the unthreading by state-of-the-art DFT-based molecular-mechanics models and a string method for the simulation of rare events. This approach enabled us to characterize in detail the unthreading mechanism, which involves the folding of the stopper during its passage through the wheel opening, a process that defies intuitive geometrical considerations. The conformational and energetic features of the transition allowed us to infer the molecular residues controlling the disassembly timescale.
Co-reporter:Marek Masnyk, Aleksandra Butkiewicz, Marcin Górecki, Roman Luboradzki, Christoph Bannwarth, Stefan Grimme, and Jadwiga Frelek
The Journal of Organic Chemistry 2016 Volume 81(Issue 11) pp:4588-4600
Publication Date(Web):April 26, 2016
DOI:10.1021/acs.joc.6b00416
The aim of the present work is to explain the causes of the observed deviations from sector and helicity rules to determine the absolute configuration of optically active α,β-unsaturated ketones by means of electronic circular dichroism (ECD). To this end, a series of model compounds with a common decahydronaphthalene skeleton representing both cisoid and transoid enones were synthesized. In the framework of this work, detailed dichroic studies supported by single crystal X-ray analysis were performed where possible. To assist the achievement of the desired objectives the conformational flexibility of the selected cis-enones through the dependence of solvent and temperature on the ECD spectra were examined. All experimental studies were supplemented by detailed DFT calculations. A notable result of the study is assessing the applicability of the enone sector and helicity rules in dichroic studies and potential restrictions. To this end, a number of factors that could determine the signs of the individual Cotton effects has been considered. Among these nonminimum structure effects, i.e., twisting of the enone chromophore and nonplanarity of the enone double bond can be mentioned.
Co-reporter:Wolfram Ratzke, Lisa Schmitt, Hideto Matsuoka, Christoph Bannwarth, Marius Retegan, Sebastian Bange, Philippe Klemm, Frank Neese, Stefan Grimme, Olav Schiemann, John M. Lupton, and Sigurd Höger
The Journal of Physical Chemistry Letters 2016 Volume 7(Issue 22) pp:4802-4808
Publication Date(Web):October 27, 2016
DOI:10.1021/acs.jpclett.6b01907
Metal-free dual singlet–triplet organic light-emitting diode (OLED) emitters can provide direct insight into spin statistics, spin correlations and spin relaxation phenomena, through a comparison of fluorescence to phosphorescence intensity. Remarkably, such materials can also function at room temperature, exhibiting phosphorescence lifetimes of several milliseconds. Using electroluminescence, quantum chemistry, and electron paramagnetic resonance spectroscopy, we investigate the effect of the conjugation pathway on radiative and nonradiative relaxation of the triplet state in phenazine-based compounds and demonstrate that the contribution of the phenazine nπ* excited state is crucial to enabling phosphorescence.
Co-reporter:Christoph Bannwarth;Jakob Seibert
Chirality 2016 Volume 28( Issue 5) pp:365-369
Publication Date(Web):
DOI:10.1002/chir.22594
Abstract
The electronic circular dichroism (ECD) spectrum of the recently synthesized [16]helicene and a derivative comprising two triisopropylsilyloxy protection groups was computed by means of the very efficient simplified time-dependent density functional theory (sTD-DFT) approach. Different from many previous ECD studies of helicenes, nonequilibrium structure effects were accounted for by computing ECD spectra on "snapshots" obtained from a molecular dynamics (MD) simulation including solvent molecules. The trajectories are based on a molecule specific classical potential as obtained from the recently developed quantum chemically derived force field (QMDFF) scheme. The reduced computational cost in the MD simulation due to the use of the QMDFF (compared to ab-initio MD) as well as the sTD-DFT approach make realistic spectral simulations feasible for these compounds that comprise more than 100 atoms. While the ECD spectra of [16]helicene and its derivative computed vertically on the respective gas phase, equilibrium geometries show noticeable differences, these are “washed” out when nonequilibrium structures are taken into account. The computed spectra with two recommended density functionals (ωB97X and BHLYP) and extended basis sets compare very well with the experimental one. In addition we provide an estimate for the missing absolute intensities of the latter. The approach presented here could also be used in future studies to capture nonequilibrium effects, but also to systematically average ECD spectra over different conformations in more flexible molecules. Chirality 28:365–369, 2016. © 2016 Wiley Periodicals, Inc.
Co-reporter:Rebecca Sure;Dr. Jan Gerit Brenburg ; Stefan Grimme
ChemistryOpen 2016 Volume 5( Issue 2) pp:94-109
Publication Date(Web):
DOI:10.1002/open.201500192
Abstract
In quantum chemical computations the combination of Hartree–Fock or a density functional theory (DFT) approximation with relatively small atomic orbital basis sets of double-zeta quality is still widely used, for example, in the popular B3LYP/6-31G* approach. In this Review, we critically analyze the two main sources of error in such computations, that is, the basis set superposition error on the one hand and the missing London dispersion interactions on the other. We review various strategies to correct those errors and present exemplary calculations on mainly noncovalently bound systems of widely varying size. Energies and geometries of small dimers, large supramolecular complexes, and molecular crystals are covered. We conclude that it is not justified to rely on fortunate error compensation, as the main inconsistencies can be cured by modern correction schemes which clearly outperform the plain mean-field methods.
Co-reporter:Tobias Heurich;Dr. Zheng-Wang Qu;Dr. Senada No&x17e;inovi&x107;;Dr. Gregor Schnakenburg;Dr. Hideto Matsuoka;Dr. Stefan Grimme;Dr. Olav Schiemann;Dr. Rainer Streubel
Chemistry - A European Journal 2016 Volume 22( Issue 29) pp:10102-10110
Publication Date(Web):
DOI:10.1002/chem.201504900
Abstract
Low-temperature generation of P-nitroxyl phosphane 2 (Ph2POTEMP), which was obtained by the reaction of Ph2PH (1) with two equivalents of TEMPO, is presented. Upon warming, phosphane 2 decomposed to give P-nitroxyl phosphane P-oxide 3 (Ph2P(O)OTEMP) as one of the final products. This facile synthetic protocol also enabled access to P-sulfide and P-borane derivatives 7 and 13, respectively, by using Ph2P(S)H (6) or Ph2P(BH3)H (11) and TEMPO. Phosphane sulfide 7 revealed a rearrangement to phosphane oxide 8 (Ph2P(O)STEMP) in CDCl3 at ambient temperature, whereas in THF, thermal decomposition of sulfide 7 yielded salt 10 ([TEMP-H2][Ph2P(S)O]). As well as EPR and detailed NMR kinetic studies, indepth theoretical studies provided an insight into the reaction pathways and spin-density distributions of the reactive intermediates.
Co-reporter:Sergej Tamke;Dr. Zheng-Wang Qu;Nikolai A. Sitte;Dr. Ulrich Flörke;Dr. Stefan Grimme;Dr. Jan Paradies
Angewandte Chemie 2016 Volume 128( Issue 13) pp:4408-4411
Publication Date(Web):
DOI:10.1002/ange.201511921
Abstract
Die erste durch ein frustriertes Lewis-Paar katalysierte Cycloisomerisierung einer Reihe von 1,5-Eninen wurde entwickelt. Die Reaktion verläuft über die π-Aktivierung des Alkins mit anschließender 5-endo-dig-Cyclisierung. Die Gegenwart von PPh3 als Lewis-Base war von besonderer Bedeutung, um einerseits Nebenreaktionen zu unterdrücken (z. B. 1,1-Carboborierung) und um andererseits die Protodeborylierung für die katalytische Reaktion zu erreichen. Der Mechanismus wurde durch quantenmechanische Rechnungen untersucht und ist im Einklang mit strukturchemischen und kinetischen Daten.
Co-reporter:Christopher Schweez;Dr. Philip Shushkov;Dr. Stefan Grimme;Dr. Sigurd Höger
Angewandte Chemie International Edition 2016 Volume 55( Issue 10) pp:3328-3333
Publication Date(Web):
DOI:10.1002/anie.201509702
Abstract
Phenylacetylene-based [2]rotaxanes were synthesized by a covalent-template approach by aminolysis of the corresponding prerotaxanes. The wheel and the bulky stoppers are made of phenylene–ethynylene–butadiynylene macrocycles of the same size. The stoppers are large enough to enable the synthesis and purification of the rotaxane. However, the wheel unthreads from the axle at elevated temperatures. The deslipping kinetics and the activation parameters were determined. We described theoretically the unthreading by state-of-the-art DFT-based molecular-mechanics models and a string method for the simulation of rare events. This approach enabled us to characterize in detail the unthreading mechanism, which involves the folding of the stopper during its passage through the wheel opening, a process that defies intuitive geometrical considerations. The conformational and energetic features of the transition allowed us to infer the molecular residues controlling the disassembly timescale.
Co-reporter:Sergej Tamke;Dr. Zheng-Wang Qu;Nikolai A. Sitte;Dr. Ulrich Flörke;Dr. Stefan Grimme;Dr. Jan Paradies
Angewandte Chemie International Edition 2016 Volume 55( Issue 13) pp:4336-4339
Publication Date(Web):
DOI:10.1002/anie.201511921
Abstract
The first frustrated Lewis pair-catalyzed cycloisomerization of a series of 1,5-enynes was developed. The reaction proceeds via the π-activation of the alkyne and subsequent 5-endo-dig cyclization with the adjacent alkene. The presence of PPh3 was of utmost importance on the one hand to prevent side reactions (for example, 1,1-carboboration) and on the other hand for the efficient protodeborylation to achieve the catalytic turnover. The mechanism is explained on the basis of quantum-chemical calculations, which are in full agreement with the experimental observations.
Co-reporter:Christoph Alexander Bauer and Stefan Grimme
The Journal of Physical Chemistry A 2016 Volume 120(Issue 21) pp:3755-3766
Publication Date(Web):May 3, 2016
DOI:10.1021/acs.jpca.6b02907
The prediction of electron ionization (EI) mass spectra (MS) from first principles has been a major challenge for quantum chemistry (QC). The unimolecular reaction space grows rapidly with increasing molecular size. On the one hand, statistical models like Eyring’s quasi-equilibrium theory and Rice–Ramsperger–Kassel–Marcus theory have provided valuable insight, and some predictions and quantitative results can be obtained from such calculations. On the other hand, molecular dynamics-based methods are able to explore automatically the energetically available regions of phase space and thus yield reaction paths in an unbiased way. We describe in this feature article the status of both methodologies in relation to mass spectrometry for small to medium sized molecules. We further present results obtained with the QCEIMS program developed in our laboratory. Our method, which incorporates stochastic and dynamic elements, has been a significant step toward the reliable routine calculation of EI mass spectra.
Co-reporter:Jens Antony, Rebecca Sure and Stefan Grimme
Chemical Communications 2015 vol. 51(Issue 10) pp:1764-1774
Publication Date(Web):14 Nov 2014
DOI:10.1039/C4CC06722C
A recently published theoretical approach employing a nondynamic structure model using dispersion-corrected density functional theory (DFT-D3) to calculate equilibrium free energies of association (Chem. Eur. J., 2012, 18, 9955–9964) is illustrated by its application to eight new supramolecular complexes. We compare with experimentally known binding constants which span the range from −3.3 to −20.3 kcal mol−1. The mean deviation of calculated from measured ΔGa results in 0.4 kcal mol−1, the mean absolute deviation in 1.8 kcal mol−1 excluding two outliers for which the computed solvation free energies are identified as the largest error source. A survey of previous applications of the theoretical approach and related computational studies is given underlining its good accuracy. It is concluded that structures and gas phase interaction energies can be computed routinely with good to high accuracy (relative errors for interaction energies of 5–10%) for complexes with about 200–300 atoms.
Co-reporter:Rebecca Sure and Stefan Grimme
Journal of Chemical Theory and Computation 2015 Volume 11(Issue 8) pp:3785-3801
Publication Date(Web):June 19, 2015
DOI:10.1021/acs.jctc.5b00296
The S12L test set for supramolecular Gibbs free energies of association ΔGa (Grimme, S. Chem. Eur. J. 2012, 18, 9955−9964) is extended to 30 complexes (S30L), featuring more diverse interaction motifs, anions, and higher charges (−1 up to +4) as well as larger systems with up to 200 atoms. Various typical noncovalent interactions like hydrogen and halogen bonding, π–π stacking, nonpolar dispersion, and CH−π and cation–dipolar interactions are represented by “real” complexes. The experimental Gibbs free energies of association (ΔGaexp) cover a wide range from −0.7 to −24.7 kcal mol–1. In order to obtain a theoretical best estimate for ΔGa, we test various dispersion corrected density functionals in combination with quadruple-ζ basis sets for calculating the association energies in the gas phase. Further, modern semiempirical methods are employed to obtain the thermostatistical corrections from energy to Gibbs free energy, and the COSMO-RS model with several parametrizations as well as the SMD model are used to include solvation contributions. We investigate the effect of including counterions for the charged systems (S30L-CI), which is found to overall improve the results. Our best method combination consists of PW6B95-D3 (for neutral and charged systems) or ωB97X-D3 (for systems with counterions) energies, HF-3c thermostatistical corrections, and Gibbs free energies of solvation obtained with the COSMO-RS 2012 parameters for nonpolar solvents and 2013-fine for water. This combination gives a mean absolute deviation for ΔGa of only 2.4 kcal mol–1 (S30L) and 2.1 kcal mol–1 (S30L-CI), with a mean deviation of almost zero compared to experiment. Regarding the relative Gibbs free energies of association for the 13 pairs of complexes which share the same host, the correct trend in binding affinities could be reproduced except for two cases. The MAD compared to experiment amounts to 1.2 kcal mol–1, and the MD is almost zero. The best-estimate theoretical corrections are used to back-correct the experimental ΔGa values in order to get an empirical estimate for the “experimental”, zero-point vibrational energy exclusive, gas phase binding energies. These are then utilized to benchmark the performance of various “low-cost” quantum chemical methods for noncovalent interactions in large systems. The performance of other common DFT methods as well as the use of semiempirical methods for structure optimizations is discussed.
Co-reporter:Holger Kruse, Arnost Mladek, Konstantinos Gkionis, Andreas Hansen, Stefan Grimme, and Jiri Sponer
Journal of Chemical Theory and Computation 2015 Volume 11(Issue 10) pp:4972-4991
Publication Date(Web):September 2, 2015
DOI:10.1021/acs.jctc.5b00515
We have created a benchmark set of quantum chemical structure–energy data denoted as UpU46, which consists of 46 uracil dinucleotides (UpU), representing all known 46 RNA backbone conformational families. Penalty-function-based restrained optimizations with COSMO TPSS-D3/def2-TZVP ensure a balance between keeping the target conformation and geometry relaxation. The backbone geometries are close to the clustering-means of their respective RNA bioinformatics family classification. High-level wave function methods (DLPNO–CCSD(T) as reference) and a wide-range of dispersion-corrected or inclusive DFT methods (DFT-D3, VV10, LC-BOP-LRD, M06-2X, M11, and more) are used to evaluate the conformational energies. The results are compared to the Amber RNA bsc0χOL3 force field. Most dispersion-corrected DFT methods surpass the Amber force field significantly in accuracy and yield mean absolute deviations (MADs) for relative conformational energies of ∼0.4–0.6 kcal/mol. Double-hybrid density functionals represent the most accurate class of density functionals. Low-cost quantum chemical methods such as PM6-D3H+, HF-3c, DFTB3-D3, as well as small basis set calculations corrected for basis set superposition errors (BSSEs) by the gCP procedure are also tested. Unfortunately, the presently available low-cost methods are struggling to describe the UpU conformational energies with satisfactory accuracy. The UpU46 benchmark is an ideal test for benchmarking and development of fast methods to describe nucleic acids, including force fields.
Co-reporter:Zheng-wang Qu, Andreas Hansen, and Stefan Grimme
Journal of Chemical Theory and Computation 2015 Volume 11(Issue 3) pp:1037-1045
Publication Date(Web):February 17, 2015
DOI:10.1021/acs.jctc.5b00007
Accurate Co–C bond dissociation energies (BDEs) of large cobalamin derivatives in the gas phase and solution are crucial for understanding bond activation mechanisms in various enzymatic reactions. However, they are challenging for both experiment and theory as indicated by an obvious discrepancy between experimental and theoretical gas phase data for adenosylcobinamide. State-of-the-art dispersion-corrected DFT and LPNO-CCSD calculations are conducted for the Co–C BDEs of some neutral and positively charged cobalamin derivatives with adenosyl and methyl ligands and compared with available experimental gas phase and solution data to resolve the controversy. Our results from various levels of electronic structure theory are fully consistent with chemical and physical reasoning. We show undoubtedly that the Co–C bonds in complexes with the bulky adenosyl ligand are indirectly enhanced by many ligand-host noncovalent interactions and that the overall BDE are larger than those with the small methyl ligand in the gas phase. The additional intramolecular dispersion and hydrogen-bond interactions are significantly but not fully quenched in aqueous solution. The theoretical results including standard continuum solvation and dispersion corrections to DFT are in full accordance with experimental solution data. This is in agreement with several successful joined experimental/theoretical studies in recent years employing similar quantum chemical methodology. We see therefore no empirical basis for questioning the reliability of current dispersion corrections like D3 or VV10 to standard density functional approximations neither for these compounds nor for organometallic chemistry in general.
Co-reporter:Christoph Bannwarth;Andreas Hansen
Israel Journal of Chemistry 2015 Volume 55( Issue 2) pp:235-242
Publication Date(Web):
DOI:10.1002/ijch.201400138
Abstract
State-of-the-art quantum chemical methods have been applied to describe the association of two frustrated Lewis pairs (FLPs), B(C6F5)3/PR3 (1: R=2,4,6-Me3C6H2; 2: R=CMe3), with different steric demands of the base component. Interaction energies are calculated at the dispersion-corrected DFT, MP2 (second-order Møller-Plesset), and DLPNO-CCSD(T) (domain-based local pair natural orbital-coupled cluster, including single, double and perturbative triple excitations) levels of theory, combined with extended triple- or quadruple-ζ AO (atom-centered orbital) basis sets. Thermostatistical contributions to the free binding energy are calculated from harmonic frequencies at the efficient HF-3c (minimal basis Hartree-Fock with three corrections) level, while solvation effects in benzene are accounted for by the COSMO-RS (conductor-like screening model for realistic solvents) continuum model. Comparison with the recently measured experimental value for the free association energy of the FLP 1 reveals agreement between theory and experiment within the estimated error bars. The computed gas phase interaction energies for both FLPs are similar (about −13 kcal mol−1), with only small variations (about ±3 kcal mol−1) for various quantum chemical methods, when London dispersion interactions are accounted for properly. The association of the more “frustrated” FLP 1 is mainly driven by nondirectional dispersion forces, resulting in non-preferential orientations, which is in agreement with experimental results. On the other hand, in FLP 2 with the “smaller” base, the boron and phosphorous atoms face each other in the favored complex structure, indicating a weak PB donor-acceptor interaction. This conformation of 2 seems to be more suitable for small molecule (e.g., H2) activations.
Co-reporter:Dr. Indranil Chatterjee;Dr. Zheng-Wang Qu;Dr. Stefan Grimme;Dr. Martin Oestreich
Angewandte Chemie 2015 Volume 127( Issue 41) pp:12326-12330
Publication Date(Web):
DOI:10.1002/ange.201504941
Abstract
Es wird über eine übergangsmetallfreie Transferhydrierung von 1,1-disubstituierten Alkenen berichtet, in der ein Cyclohexa-1,4-dien die formale Diwasserstoffquelle darstellt. Der Prozess beginnt mit einer B(C6F5)3-vermittelten Hydridabstraktion vom Diwasserstoffsurrogat unter Bildung eines Brønsted-sauren Wheland-Komplexes sowie [HB(C6F5)3]−. Eine Abfolge aus Protonen- und Hydridübertragungen auf das Alkensubstrat liefert dann das Alkan. Obwohl mehrere Carbeniumionzwischenstufen beteiligt sind, werden konkurrierende Reaktionswege, beispielsweise Diwasserstofffreisetzung und kationische Dimerisierungen von Reaktanten, weitgehend unterdrückt. Dies wird durch den Einsatz von Cyclohexa-1,4-dienen mit Methylgruppen an den C1- und C5-Positionen sowie am C3-Kohlenstoffatom, an dem die Hydridabstraktion stattfindet, ermöglicht. Die Konzentration an Alken ist eine weitere entscheidende Einflussgröße. Die unterschiedlichen Reaktionspfade wurden berechnet, woraus sich ein mechanistisches Bild ergibt, das mit den experimentellen Beobachtungen vollständig in Einklang ist.
Co-reporter:Dr. Indranil Chatterjee;Dr. Zheng-Wang Qu;Dr. Stefan Grimme;Dr. Martin Oestreich
Angewandte Chemie International Edition 2015 Volume 54( Issue 41) pp:12158-12162
Publication Date(Web):
DOI:10.1002/anie.201504941
Abstract
A transition-metal-free transfer hydrogenation of 1,1-disubstituted alkenes with cyclohexa-1,4-dienes as the formal source of dihydrogen is reported. The process is initiated by B(C6F5)3-mediated hydride abstraction from the dihydrogen surrogate, forming a Brønsted acidic Wheland complex and [HB(C6F5)3]−. A sequence of proton and hydride transfers onto the alkene substrate then yields the alkane. Although several carbenium ion intermediates are involved, competing reaction channels, such as dihydrogen release and cationic dimerization of reactants, are largely suppressed by the use of a cyclohexa-1,4-diene with methyl groups at the C1 and C5 as well as at the C3 position, the site of hydride abstraction. The alkene concentration is another crucial factor. The various reaction pathways were computationally analyzed, leading to a mechanistic picture that is in full agreement with the experimental observations.
Co-reporter:Christoph Bannwarth and Stefan Grimme
The Journal of Physical Chemistry A 2015 Volume 119(Issue 15) pp:3653-3662
Publication Date(Web):March 23, 2015
DOI:10.1021/acs.jpca.5b01680
We show that the electronic circular dichroism (ECD) of delocalized π-systems represents a worst-case scenario for Tamm–Dancoff approximated (TDA) linear response methods. We mainly consider density functional theory (TDA-DFT) variants together with range-separated hybrids, but the conclusions also apply for other functionals as well as the configuration interaction singles (CIS) approaches. We study the effect of the TDA for the computation of ECD spectra in some prototypical extended π-systems. The C76 fullerene, a chiral carbon nanotube fragment, and [11]helicene serve as model systems for inherently chiral, π-chromophores. Solving the full linear response problem is inevitable in order to obtain accurate ECD spectra for these systems. For the C76 fullerene and the nanotube fragment, TDA and CIS approximated methods yield spectra in the origin-independent velocity gauge formalism of incorrect sign which would lead to the assignment of the opposite (wrong) absolute configuration. As a counterexample, we study the ECD of an α-helix polypeptide chain. Here, the lowest-energy transitions are dominated by localized excitations within the individual peptide units, and TDA methods perform satisfactorily. The results may have far-reaching implications for simple semiempirical methods which often employ TDA and CIS for huge molecules. Our recently presented simplified time-dependent DFT approach proves to be an excellent low-cost linear response method which together with range-separated density functionals like ωB97X-D3 produces ECD spectra in very good agreement with experiment.
Co-reporter:Dr. Stefan Grimme;Dr. Andreas Hansen
Angewandte Chemie International Edition 2015 Volume 54( Issue 42) pp:12308-12313
Publication Date(Web):
DOI:10.1002/anie.201501887
Abstract
The inclusion of dynamical and static electron correlation (SEC) is mandatory for accurate quantum chemistry (QC). SEC is particularly difficult to calculate and hence a qualitative understanding is important to judge the applicability of approximate QC methods. Existing scalar SEC diagnostics, however, lack the important information where the SEC effects occur in a molecule. We introduce an analysis tool based on a fractional occupation number weighted electron density (ρFOD) that is plotted in 3D for a pre-defined contour surface value. The scalar field is obtained by finite-temperature DFT calculations with pre-defined electronic temperature (e.g. TPSS at 5000 K). FOD plots only show the contribution of the “hot” (strongly correlated) electrons. We discuss illustrative plots for a broad range of chemical systems from small molecules to large conjugated molecules with polyradicaloid character. Spatial integration yields a single number which can be used to globally quantify SEC.
Co-reporter:Dr. Stefan Grimme;Dr. Andreas Hansen
Angewandte Chemie 2015 Volume 127( Issue 42) pp:12483-12488
Publication Date(Web):
DOI:10.1002/ange.201501887
Abstract
Die Einbeziehung von dynamischer und statischer Elektronenkorrelation (SEK) ist unerlässlich für hochgenaue quantenchemische (QC) Berechnungen, wobei die SEK besonders schwierig zu beschreiben ist. Daher ist ein qualitatives Verständnis wichtig, um die Anwendbarkeit von approximativen QC-Methoden zu beurteilen. Bereits vorhandenen SEK-Diagnostiken fehlt jedoch die wichtige Information, wo in einem Molekül SEK-Effekte auftreten. Wir stellen ein Analysewerkzeug vor, welches auf einer mit gebrochenzahlig besetzten Orbitalen gewichteten Elektronendichte (ρFOD, “fractional occupation density”) basiert und in 3D mit einem vordefinierten Konturwert als Isofläche dargestellt wird. Dieses Skalarfeld wird aus einer Dichtefunktionaltheorie(DFT)-Rechnung mit einer festen elektronischen Temperatur (z. B. TPSS mit 5000 K) erhalten. FOD-Visualisierungen zeigen nur die Beiträge “heißer” (stark korrelierter) Elektronen. Wir diskutieren FOD-Abbildungen für eine breite Palette von typischen chemischen Systemen, angefangen von kleinen Molekülen bis hin zu großen, konjugierten Molekülen mit Polyradikalcharakter. Durch räumliche Integration der ρFOD erhält man eine Maßzahl, welche zur weiteren Quantifizierung der SEK benutzt werden kann.
Co-reporter:René Liedtke ; Felix Scheidt ; Jinjun Ren ; Birgitta Schirmer ; Allan Jay P. Cardenas ; Constantin G. Daniliuc ; Hellmut Eckert ; Timothy H. Warren ; Stefan Grimme ; Gerald Kehr ;Gerhard Erker
Journal of the American Chemical Society 2014 Volume 136(Issue 25) pp:9014-9027
Publication Date(Web):May 21, 2014
DOI:10.1021/ja5028293
The vicinal frustrated Lewis pair (FLP) mes2P–CH2CH2–B(C6F5)2 (3) reacts with phenyl(trimethylsilyl)acetylene by 1,1-carboboration to give the extended C3-bridged FLP 6 featuring a substituted vinylborane subunit. The FLP 6 actively cleaves dihydrogen. The FLP 3 also undergoes a 1,1-carboboration reaction with diphenylphosphino(trimethylsilyl)acetylene to give the P/B/P FLP 11 that features a central unsaturated four-membered heterocyclic P/B FLP and a pendant CH2CH2–Pmes2 functional group. Compound 11 reacts with nitric oxide (NO) by oxidation of the pendant Pmes2 unit to the P(O)mes2 phosphine oxide and N,N-addition of the P/B FLP unit to NO to yield the persistent P/B/PO FLPNO aminoxyl radical 14. This reaction is initiated by P(O)mes2 formation and opening of the central Ph2P···B(C6F5)2 linkage triggered by the pendant CH2CH2–P(O)mes2 group.
Co-reporter:Christoph Gütz ; Rainer Hovorka ; Niklas Struch ; Jens Bunzen ; Georg Meyer-Eppler ; Zheng-Wang Qu ; Stefan Grimme ; Filip Topić ; Kari Rissanen ; Mario Cetina ; Marianne Engeser ;Arne Lützen
Journal of the American Chemical Society 2014 Volume 136(Issue 33) pp:11830-11838
Publication Date(Web):July 22, 2014
DOI:10.1021/ja506327c
A tris(bipyridine) ligand 1 with two BINOL (BINOL = 2,2′-dihydroxy-1,1′-binaphthyl) groups has been prepared in two enantiomerically pure forms. This ligand undergoes completely diastereoselective self-assembly into D2-symmeteric double-stranded trinuclear helicates upon coordination to copper(I) and silver(I) ions and to D3-symmetric triple-stranded trinuclear helicates upon coordination to copper(II), zinc(II), and iron(II) ions as demonstrated by mass spectrometry, NMR and CD spectroscopy in combination with quantum chemical calculations and X-ray diffraction analysis. According to the calculations, the single diastereomers that are formed during the self-assembly process are strongly preferred compared to the next stable diastereomers. Due to this strong preference, the self-assembly of the helicates from racemic 1 proceeds in a completely narcissistic self-sorting manner with an extraordinary high degree of self-sorting that proves the power and reliability of this approach to achieve high-fidelity diastereoselective self-assembly via chiral self-sorting to get access to stereochemically well-defined nanoscaled objects. Furthermore, mass spectrometric methods including electron capture dissociation MSn experiments could be used to elucidate the redox behavior of the copper helicates.
Co-reporter:David Schweinfurth, Serhiy Demeshko, Stephan Hohloch, Marc Steinmetz, Jan Gerit Brandenburg, Sebastian Dechert, Franc Meyer, Stefan Grimme, and Biprajit Sarkar
Inorganic Chemistry 2014 Volume 53(Issue 16) pp:8203-8212
Publication Date(Web):August 4, 2014
DOI:10.1021/ic500264k
The complexes [Fe(tbta)2](BF4)2·2EtOH (1), [Fe(tbta)2](BF4)2·2CH3CN (2), [Fe(tbta)2](BF4)2·2CHCl3 (3), and [Fe(tbta)2](BF4)2 (4) were synthesized from the respective metal salts and the click-derived tripodal ligand tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine (tbta). Structural characterization of these complexes (at 100 or 133 K) revealed Fe–N bond lengths for the solvent containing compounds 1–3 that are typical of a high spin (HS) Fe(II) complex. In contrast, the solvent-free compound 4 show Fe–N bond lengths that are characteristic of a low spin (LS) Fe(II) state. The Fe center in all complexes is bound to two triazole and one amine N atom from each tbta ligand, with the third triazole arm remaining uncoordinated. The benzyl substituents of the uncoordinated triazole arms and the triazole rings engage in strong intermolecular and intramolecular noncovalent interactions. These interactions are missing in the solvent containing molecules 1, 2, and 3, where the solvent molecules occupy positions that hinder these noncovalent interactions. The solvent-free complex (4) displays spin crossover (SCO) with a spin transition temperature T1/2 near room temperature, as revealed by superconducting quantum interference device (SQUID) magnetometric and Mössbauer spectroscopic measurements. The complexes 1, 2, and 3 remain HS throughout the investigated temperature range. Different torsion angles at the metal centers, which are influenced by the noncovalent interactions, are likely responsible for the differences in the magnetic behavior of these complexes. The corresponding solvent-free Co(II) complex (6) is also LS at lower temperatures and displays SCO with a temperature T1/2 near room temperature. Theoretical calculations at molecular and periodic DFT-D3 levels for 1–4 qualitatively reproduce the experimental findings, and corroborate the importance of intermolecular and intramolecular noncovalent interactions for the magnetic properties of these complexes. The present work thus represents rare examples of SCO complexes where the use of identical ligand sets produces SCO in Fe(II) as well as Co(II) complexes.
Co-reporter:Stefan Grimme
Journal of Chemical Theory and Computation 2014 Volume 10(Issue 10) pp:4497-4514
Publication Date(Web):August 25, 2014
DOI:10.1021/ct500573f
A black-box type procedure is presented for the generation of molecule-specific, classical potential energy functions (force-field, FF) solely from quantum mechanically (QM) computed input data. The approach can treat covalently bound molecules and noncovalent complexes with almost arbitrary structure. The necessary QM information consists of the equilibrium structure and the corresponding Hessian matrix, atomic partial charges, and covalent bond orders. The FF fit is performed automatically without any further input and yields a specific (nontransferable) potential which very closely resembles the QM reference potential near the equilibrium. The resulting atomistic, fully flexible FF is anharmonic and allows smooth dissociation of covalent bonds into atoms. A newly proposed force-constant–bond-energy relation with little empiricism provides reasonably accurate (about 5–10% error) atomization energies for almost arbitrary diatomic and polyatomic molecules. Intra- and intermolecular noncovalent interactions are treated by using well established and accurate D3 dispersion coefficients, CM5 charges from small basis set QM calculations, and a new interatomic repulsion potential. Particular attention has been paid to the construction of the torsion potentials which are partially obtained from automatic QM-tight-binding calculations for model systems. Detailed benchmarks are presented for conformational energies, atomization energies, vibrational frequencies, gas phase structures of organic molecules, and transition metal complexes. Comparisons to results from standard FF or semiempirical methods reveal very good accuracy of the new potential. While further studies are necessary to validate the approach, the initial results suggest QMDFF as a routine tool for the computation of a wide range of properties and systems (e.g., for molecular dynamics of isolated molecules, explicit solvation, self-solvation (melting) or even for molecular crystals) in particular when standard parametrizations are unavailable.
Co-reporter:Christoph Alexander Bauer and Stefan Grimme
Organic & Biomolecular Chemistry 2014 vol. 12(Issue 43) pp:8737-8744
Publication Date(Web):08 Sep 2014
DOI:10.1039/C4OB01668H
This study presents a showcase for the novel Quantum Chemistry Electron Ionization Mass Spectrometry (QCEIMS) method on five FDA-approved drugs. The method allows a first-principles electronic structure-based prediction of EI mass spectra in principle for any molecule. The systems in this case study are organic substances of nominal masses between 404 and 853 atomic mass units and cover a wide range of functional groups and organic molecular structure motifs. The results demonstrate the widespread applicability of the QCEIMS method for the unbiased computation of EI mass spectra even for larger molecules. Its strengths compared to standard (static) or database driven approaches in such cases are highlighted. Weak points regarding the required computation times or the approximate character of the employed QC methods are also discussed. We propose QCEIMS as a viable and robust way of predicting EI mass spectra for sizeable organic molecules relevant to medicinal and pharmaceutical chemistry.
Co-reporter:Predrag V. Petrović, Stefan Grimme, Snežana D. Zarić, Michel Pfeffer and Jean-Pierre Djukic
Physical Chemistry Chemical Physics 2014 vol. 16(Issue 28) pp:14688-14698
Publication Date(Web):02 Jun 2014
DOI:10.1039/C4CP01500B
Self-aggregation in water of anti-cancer agents such as oxaliplatin (1) or its palladium-containing parent (2) is suspected to be the main reason for the exceptional resistance of concentrated infusions of these complexes to hydrolysis; this hypothesis, i.e. the self-association of metal chelates, was investigated in a systematic manner by experimental and theoretical means. 1H diffusion-ordered NMR spectroscopy (DOSY NMR) and UV-visible absorption titration were inconclusive as to the formation of a dimer of 1 in water or DMSO. Further isothermal titration calorimetry (ITC) methods allowed the accurate determination of the enthalpy of formation of only the homodimer [2]2 and putative heterodimer [1·2] together with an estimation of the formation constants, which indicate that dimer formation is not a spontaneous process in solution, whereas electrospray ESI mass spectroscopy tends to suggest the contrary in the gas phase. A dispersion-corrected DFT method, i.e. DFT-D (BLYP-D3), was used to model the aggregation in solution (COSMO) and to investigate the assisting role of London force in the cohesion of bimolecular aggregates. The concordance of experimental and theoretical thermodynamic parameters was judged reasonably even though the treatment of solvation by conventional continuum models does not account for specific interactions of the solute with molecules of solvent; nonetheless these results outline the importance of dispersion, a.k.a. London force. The role of the latter was further stressed by computing the affinities of 1 and 2 for the lipophilic cavity of cucurbit[7]uril in modeled water (COSMO-RS), which were preliminarily determined experimentally by ITC methods using pure water as solvent. From our investigations carried out in pure water the connection between the notorious chemical stability of “concentrated” infusions of 1 in aqueous media and the formation of oligomers remains unsettled.
Co-reporter:Tobias Risthaus, Andreas Hansen and Stefan Grimme
Physical Chemistry Chemical Physics 2014 vol. 16(Issue 28) pp:14408-14419
Publication Date(Web):29 Nov 2013
DOI:10.1039/C3CP54517B
The recently introduced sTDA methodology [S. Grimme, J. Chem. Phys., 2013, 138, 244104] to compute excitation spectra of huge molecular systems is extended to range-separated hybrid (RSH) density functionals. The three empirical parameters of the method which describe a screened two-electron interaction are obtained for some common RSH functionals (ωB97 family, CAM-B3LYP, LC-BLYP) from a fit to theoretical SCS-CC2 reference vertical excitation energies for a set of small to medium-sized chromophores. The method is cross-validated on a set of inter- and intramolecular charge transfer states and a set composed of typical valence transitions. Overall small deviations from reference data of only about 0.2–0.4 eV are found with best performance for CAM-B3LYP and ωB97X-D3. To demonstrate versatility and robustness of the new methodology, applications (the UV/Vis spectrum of the pyridine polymer and the ECD spectrum of (P)-[11]helicene) and frequently used charge transfer examples are discussed. In one case, 11000+ excited electronic states of a system containing 330 atoms were calculated. We show that the asymptotically correct sTDA–RSH combination yields results often superior to those based on global hybrids and that it opens up new possibilities for the computation of excited states in materials science and bio-molecular systems.
Co-reporter:Rebecca Sure, Jens Antony, and Stefan Grimme
The Journal of Physical Chemistry B 2014 Volume 118(Issue 12) pp:3431-3440
Publication Date(Web):March 3, 2014
DOI:10.1021/jp411616b
Association free energies ΔGa are calculated for two different types of host–guest systems, the rigid cucurbit[7]uril (CB7) and the basket shaped octa-acid (OA), and a number of charged guest molecules each by quantum chemical methods from first principles in the context of a recent blind test challenge (SAMPL4). For CB7, the overall agreement between theory and experiment is excellent. In comparison with all other submitted calculated relative ΔGa,rel values for this part of the blind test, our results ranked on top. Modeling the binding free energy in the case of the OA host mainly suffers from the problem that the binding situation is undefined with respect to the charge state and due to its intrinsic flexibility the host–guest complex is not represented well by a single configuration, but qualitative features of the binding process such as the proper binding orientation and the order of magnitude of ΔGa are represented in accord with the experimental expectations even though an accurate ranking is not possible.
Co-reporter:Thomas Wiegand, Hellmut Eckert, Jinjun Ren, Gunther Brunklaus, Roland Fröhlich, Constantin G. Daniliuc, Gerrit Lübbe, Kathrin Bussmann, Gerald Kehr, Gerhard Erker, and Stefan Grimme
The Journal of Physical Chemistry A 2014 Volume 118(Issue 12) pp:2316-2331
Publication Date(Web):February 21, 2014
DOI:10.1021/jp500172b
No-bond 31P–31P indirect dipolar couplings, which arise from the transmission of nuclear spin polarization through interaction of proximal nonbonded electron pairs have been investigated in the solid state for a series of closely related substituted P,P-[3]ferrocenophanes and model systems. Through variation and combination of ligands (phenyl, cyclohexyl, isopropyl) at the two phosphorus sites, the P···P distances in these compounds can be varied from 3.49 to 4.06 Å. Thus, the distance dependence of the indirect no-bond coupling constant Jnb can be studied in a series of closely related compounds. One- and two-dimensional solid-state NMR experiments serve to establish the character of these couplings and to measure the isotropic coupling constants Jiso, which were found to range between 12 and 250 Hz. To develop an understanding of the magnitude of Jnb in terms of molecular structure, their dependences on intramolecular internuclear distances and relative orbital orientations is discussed by DFT-calculations on suitable models. In agreement with the literature the dependence of Jnb on the P···P distance is found to be exponential; however, the steepness of this curve is highly dependent on the internuclear equilibrium distance. For a quantitative description, the off-diagonal elements of the expectation value of the Kohn–Sham–Fock operator in the LMO basis for the LMOs of the two phosphorus lone-pairs is proposed. This parameter correlates linearly with the calculated Jnb values and possesses the same distance-dependence. In addition, the simulations indicate a distinct dependence of Jnb on the dihedral angle defined by the two C–P bonds providing ligation to the molecular backbone. For disordered materials or those featuring multiple sites, conformers, and/or polymorphism, a new double-quantum NMR method termed DQ-DRENAR can be used to conveniently measure internuclear 31P–31P distances. If conducted on compounds with known P···P distances, such measurements can also serve to estimate the magnitude of the anisotropy ΔJ of these no-bond indirect spin–spin couplings. The DFT results suggest that in the present series of compounds the magnitude of ΔJ is strongly correlated with that of the isotropic component, as both parameters have analogous distance dependences. While our studies indicate a sizable J-anisotropy for the model compound 1,8-bis(diphenylphosphino)napthalene (ΔJ ∼ −70 Hz), the corresponding values for the P,P-[3]ferrocenophanes are significantly smaller, affecting their DQ-DRENAR curves only in a minor way. Based on the above insights, the structural aspects of conformational disorder and polymorphism observed in some of the P,P-[3]ferrocenophanes are discussed.
Co-reporter:Christoph Alexander Bauer and Stefan Grimme
The Journal of Physical Chemistry A 2014 Volume 118(Issue 49) pp:11479-11484
Publication Date(Web):November 13, 2014
DOI:10.1021/jp5096618
The gas phase fragmentation pathways of the nucleobase adenine upon 70 eV electron ionization are investigated by means of a combined stochastic and first-principles based molecular dynamics approach. We employ no preconceived fragmentation channels in our calculations, which simulate standard electron ionization mass spectrometry (EI-MS) conditions. The reactions observed compare well to a wealth of experimental and theoretical data available for this important nucleic acid building block. All significant peaks in the experimental mass spectrum of adenine are reproduced. Additionally, the fragment ion connectivities obtained from our simulations at least partially concur with results from previous experimental studies on selectively isotope labeled adenines. Moreover, we are able to assign noncyclic structures that are entropically favored and have not been proposed in nondynamic quantum chemical studies before to the decomposition products, which result automatically from our molecular dynamics procedure. From simulations under various conditions it is evident that most of the fragmentation reactions even at low internal excess energy (<10 eV) occur very fast within a few picoseconds.
Co-reporter:Dr. Andreas Hansen;Christoph Bannwarth;Dr. Stefan Grimme;Predrag Petrovi&x107;;Christophe Werlé;Dr. Jean-Pierre Djukic
ChemistryOpen 2014 Volume 3( Issue 5) pp:177-189
Publication Date(Web):
DOI:10.1002/open.201402017
Abstract
Reliable thermochemical measurements and theoretical predictions for reactions involving large transition metal complexes in which long-range intramolecular London dispersion interactions contribute significantly to their stabilization are still a challenge, particularly for reactions in solution. As an illustrative and chemically important example, two reactions are investigated where a large dipalladium complex is quenched by bulky phosphane ligands (triphenylphosphane and tricyclohexylphosphane). Reaction enthalpies and Gibbs free energies were measured by isotherm titration calorimetry (ITC) and theoretically ‘back-corrected’ to yield 0 K gas-phase reaction energies (ΔE). It is shown that the Gibbs free solvation energy calculated with continuum models represents the largest source of error in theoretical thermochemistry protocols. The (‘back-corrected’) experimental reaction energies were used to benchmark (dispersion-corrected) density functional and wave function theory methods. Particularly, we investigated whether the atom-pairwise D3 dispersion correction is also accurate for transition metal chemistry, and how accurately recently developed local coupled-cluster methods describe the important long-range electron correlation contributions. Both, modern dispersion-corrected density functions (e.g., PW6B95-D3(BJ) or B3LYP-NL), as well as the now possible DLPNO-CCSD(T) calculations, are within the ‘experimental’ gas phase reference value. The remaining uncertainties of 2–3 kcal mol−1 can be essentially attributed to the solvation models. Hence, the future for accurate theoretical thermochemistry of large transition metal reactions in solution is very promising.
Co-reporter:Jan Gerit Brandenburg, Manuel Hochheim, Thomas Bredow, and Stefan Grimme
The Journal of Physical Chemistry Letters 2014 Volume 5(Issue 24) pp:4275-4284
Publication Date(Web):November 25, 2014
DOI:10.1021/jz5021313
The efficient and reasonably accurate description of noncovalent interactions is important for various areas of chemistry, ranging from supramolecular host–guest complexes and biomolecular applications to the challenging task of crystal structure prediction. While London dispersion inclusive density functional theory (DFT-D) can be applied, faster “low-cost” methods are required for large-scale applications. In this Perspective, we present the state-of-the-art of minimal basis set, semiempirical molecular-orbital-based methods. Various levels of approximations are discussed based either on canonical Hartree–Fock or on semilocal density functionals. The performance for intermolecular interactions is examined on various small to large molecular complexes and organic solids covering many different chemical groups and interaction types. We put the accuracy of low-cost methods into perspective by comparing with first-principle density functional theory results. The mean unsigned deviations of binding energies from reference data are typically 10–30%, which is only two times larger than those of DFT-D. In particular, for neutral or moderately polar systems, many of the tested methods perform very well, while at the same time, computational savings of up to 2 orders of magnitude can be achieved.
Co-reporter:Jan Gerit Brandenburg and Stefan Grimme
The Journal of Physical Chemistry Letters 2014 Volume 5(Issue 11) pp:1785-1789
Publication Date(Web):May 6, 2014
DOI:10.1021/jz500755u
The ambitious goal of organic crystal structure prediction challenges theoretical methods regarding their accuracy and efficiency. Dispersion-corrected density functional theory (DFT-D) in principle is applicable, but the computational demands, for example, to compute a huge number of polymorphs, are too high. Here, we demonstrate that this task can be carried out by a dispersion-corrected density functional tight binding (DFTB) method. The semiempirical Hamiltonian with the D3 correction can accurately and efficiently model both solid- and gas-phase inter- and intramolecular interactions at a speed up of 2 orders of magnitude compared to DFT-D. The mean absolute deviations for interaction (lattice) energies for various databases are typically 2–3 kcal/mol (10–20%), that is, only about two times larger than those for DFT-D. For zero-point phonon energies, small deviations of <0.5 kcal/mol compared to DFT-D are obtained.Keywords: dispersion correction; noncovalent interaction; organic crystals; semiempirical MO; tight binding;
Co-reporter:Jonas Moellmann
The Journal of Physical Chemistry C 2014 Volume 118(Issue 14) pp:7615-7621
Publication Date(Web):March 13, 2014
DOI:10.1021/jp501237c
We investigate the performance of the dispersion correction D3 with and without an explicit three-body dispersion term for the energetic and structural properties of rare gas and molecular crystals. Therefore, the two- and three-body gradient of the dispersion energy is implemented in the periodic plane-wave program VASP. It is combined with different density functionals at the level of the general gradient approximation (GGA) and hybrid functionals. Cohesive energies and lattice parameters for the rare gas crystals Ar, Kr, and Xe and a set of 23 molecular crystals are calculated and compared to experimental reference values. In general, all tested methods yield very good results. For the molecular crystals the mean absolute deviation of lattice energies from reference data (about 1–2 kcal/mol) is close to or below their uncertainties. The influence of the three-body Axilrod–Teller–Muto dispersion term on energy and structure is found to be rather small. While on a GGA level cohesive energies become slightly worse, for hybrid functionals the three-body term improves the results.
Co-reporter:Jan Gerit Brandenburg, Georg Bender, Jinjun Ren, Andreas Hansen, Stefan Grimme, Hellmut Eckert, Constantin G. Daniliuc, Gerald Kehr, and Gerhard Erker
Organometallics 2014 Volume 33(Issue 19) pp:5358-5364
Publication Date(Web):September 15, 2014
DOI:10.1021/om500678p
We present a combined theoretical and experimental analysis of the carbon–carbon bond character in two prototypical zirconocene complexes. The two cyclic seven-membered ring zirconium compounds 2a and 2b differ by the substitution of a tert-butyl by a trimethylsilyl group. Due to the coordination of the π-system to the metal atom, a formally forbidden (η2-allenyl)/enamido-Zr to (η2-alkyne)/κN-imine-Zr complex isomerization is feasible. State-of-the-art dispersion-corrected density functional theory (DFT-D3) is used in both the solid and condensed phase to examine and quantify the experimental structures (X-ray diffraction) and 13C NMR magnetic shielding. The complementary investigations demonstrate the importance of nonlocal London dispersion interactions. Both X-ray structures agree excellently with the results of the solid-state DFT-D3 calculations. Interestingly, 2b exhibits a mixed allene–alkyne form in the solid state, while its gas phase structure has a strong allene character. The substitution leading to 2a prevents this isomerization in the solid state by the intramolecular stabilization of the allene structure. NMR solid and liquid phase measurements confirm the theoretically proposed transition. By combining the experimental and theoretical information, the rather unusual triple/single to double/double-bond transition is attributed to an intermolecular London dispersion induced crystal packing effect.
Co-reporter:Stephan Ehrlich, Jonas Moellmann, and Stefan Grimme
Accounts of Chemical Research 2013 Volume 46(Issue 4) pp:916
Publication Date(Web):June 15, 2012
DOI:10.1021/ar3000844
Aromatic interactions play a key role in many chemical and biological systems. However, even if very simple models are chosen, the systems of interest are often too large to be handled with standard wave function theory (WFT). Although density functional theory (DFT) can easily treat systems of more than 200 atoms, standard semilocal (hybrid) density functional approximations fail to describe the London dispersion energy, a factor that is essential for accurate predictions of inter- and intramolecular noncovalent interactions. Therefore dispersion-corrected DFT provides a unique tool for the investigation and analysis of a wide range of complex aromatic systems.In this Account, we start with an analysis of the noncovalent interactions in simple model dimers of hexafluorobenzene (HFB) and benzene, with a focus on electrostatic and dispersion interactions. The minima for the parallel-displaced dimers of HFB/HFB and HFB/benzene can only be explained when taking into account all contributions to the interaction energy and not by electrostatics alone. By comparison of saturated and aromatic model complexes, we show that increased dispersion coefficients for sp2-hybridized carbon atoms play a major role in aromatic stacking.Modern dispersion-corrected DFT yields accurate results (about 5–10% error for the dimerization energy) for the relatively large porphyrin and coronene dimers, systems for which WFT can provide accurate reference data only with huge computational effort. In this example, it is also demonstrated that new nonlocal, density-dependent dispersion corrections and atom pairwise schemes mutually agree with each other.The dispersion energy is also important for the complex inter- and intramolecular interactions that arise in the molecular crystals of aromatic molecules. In studies of hexahelicene, dispersion-corrected DFT yields “the right answer for the right reason”. By comparison, standard DFT calculations reproduce intramolecular distances quite accurately in single-molecule calculations while inter- and intramolecular distances become too large when dispersion-uncorrected solid-state calculations are carried out. Dispersion-corrected DFT can fix this problem, and these results are in excellent agreement with experimental structure and energetic (sublimation) data. Uncorrected treatments do not even yield a bound crystal state.Finally, we present calculations for the formation of a cationic, quadruply charged dimer of a porphyrin derivative, a case where dispersion is required in order to overcome strong electrostatic repulsion. A combination of dispersion-corrected DFT with an adequate continuum solvation model can accurately reproduce experimental free association enthalpies in solution. As in the previous examples, consideration of the electrostatic interactions alone does not provide a qualitatively or quantitatively correct picture of the interactions of this complex.
Co-reporter:Jiří Šponer ; Arnošt Mládek ; Naďa Špačková ; Xiaohui Cang ; Thomas E. Cheatham ; III
Journal of the American Chemical Society 2013 Volume 135(Issue 26) pp:9785-9796
Publication Date(Web):June 6, 2013
DOI:10.1021/ja402525c
We provide theoretical predictions of the intrinsic stability of different arrangements of guanine quadruplex (G-DNA) stems. Most computational studies of nucleic acids have applied Molecular Mechanics (MM) approaches using simple pairwise-additive force fields. The principle limitation of such calculations is the highly approximate nature of the force fields. In this study, we for the first time apply accurate QM computations (DFT-D3 with large atomic orbital basis sets) to essentially complete DNA building blocks, seven different folds of the cation-stabilized two-quartet G-DNA stem, each having more than 250 atoms. The solvent effects are approximated by COSMO continuum solvent. We reveal sizable differences between MM and QM descriptions of relative energies of different G-DNA stems, which apparently reflect approximations of the DNA force field. Using the QM energy data, we propose correction to earlier free energy estimates of relative stabilities of different parallel, hybrid, and antiparallel G-stem folds based on classical simulations. The new energy ranking visibly improves the agreement between theory and experiment. We predict the 5′-anti-anti-3′ GpG dinucleotide step to be the most stable one, closely followed by the 5′-syn-anti-3′ step. The results are in good agreement with known experimental structures of 2-, 3-, and 4-quartet G-DNA stems. Besides providing specific results for G-DNA, our study highlights basic limitations of force field modeling of nucleic acids. Although QM computations have their own limitations, mainly the lack of conformational sampling and the approximate description of the solvent, they can substantially improve the quality of calculations currently relying exclusively on force fields.
Co-reporter:Muhammad Sajid ; Arunlibertsen Lawzer ; Weishi Dong ; Christoph Rosorius ; Wolfram Sander ; Birgitta Schirmer ; Stefan Grimme ; Constantin G. Daniliuc ; Gerald Kehr ;Gerhard Erker
Journal of the American Chemical Society 2013 Volume 135(Issue 49) pp:18567-18574
Publication Date(Web):October 22, 2013
DOI:10.1021/ja408815k
The intramolecular frustrated Lewis pair (FLP) Mes2PCH2CH2B(C6F5)2 4 adds cooperatively to carbon monoxide to form the five-membered heterocyclic carbonyl compound 5. The intramolecular FLP 7 contains an exo-3-B(C6F5)2 Lewis acid and an endo-2-PMes2 Lewis base functionality coordinated at the norbornane framework. This noninteracting FLP adds carbon monoxide in solution at −35 °C cooperatively to yield a five-membered heterocyclic FLP-carbonyl compound 8. In contrast, FLP 7 is carbonylated in a CO-doped argon matrix at 25 K to selectively form a borane carbonyl 9 without involvement of the adjacent phosphanyl moiety. The free FLP 7 was generated in the gas phase from its FLPH2 product 10. A DFT study has shown that the phosphonium hydrido borate zwitterion 10 is formed exergonically in solution but tends to lose H2 when brought into the gas phase.
Co-reporter:José Clayston Melo Pereira ; Muhammad Sajid ; Gerald Kehr ; Ashley M. Wright ; Birgitta Schirmer ; Zheng-Wang Qu ; Stefan Grimme ; Gerhard Erker ;Peter C. Ford
Journal of the American Chemical Society 2013 Volume 136(Issue 1) pp:513-519
Publication Date(Web):December 13, 2013
DOI:10.1021/ja4118335
Described is a kinetics and computational study of the reaction of NO with the intramolecular bridged P/B frustrated Lewis pair (FLP) endo-2-(dimesitylphosphino)-exo-3-bis(pentafluorophenyl)boryl-norbornane to give a persistent FLP-NO aminoxyl radical. This reaction follows a second-order rate law, first-order in [FLP] and first-order in [NO], and is markedly faster in toluene than in dichloromethane. By contrast, the NO oxidation of the phosphine base 2-(dimesitylphosphino)norbornene to the corresponding phosphine oxide follows a third-order rate law, first-order in [phosphine] and second-order in [NO]. Formation of the FLP-NO radical in toluene occurs with a ΔH⧧ of 13 kcal mol–1, a feature that conflicts with the computation-based conclusion that NO addition to a properly oriented B/P pair should be nearly barrierless. Since the calculations show the B/P pair in the most stable solution structure of this FLP to have an unfavorable orientation for concerted reaction, the observed barrier is rationalized in terms of the reversible formation of a [B]-NO complex intermediate followed by a slower isomerization–ring closure step to the cyclic aminoxyl radical. This combined kinetics/theoretical study for the first time provides insight into mechanistic details for the activation of a diatomic molecule by a prototypical FLP.
Co-reporter:Ramesh C. Samanta, Suman De Sarkar, Roland Fröhlich, Stefan Grimme and Armido Studer
Chemical Science 2013 vol. 4(Issue 5) pp:2177-2184
Publication Date(Web):05 Mar 2013
DOI:10.1039/C3SC00099K
This edge article reports the synthesis and full characterization including X-ray analysis of three different acylazolium ions. The reactivity of these acylazolium ions as acylating reagents of amines and alcohols is discussed. Whereas benzylamine slowly reacts with the acylazolium ions, benzyl alcohol acylation does not occur. However, upon activation of the alcohol with an N-heterocyclic carbene (NHC) as catalyst, efficient esterification is achieved. Importantly, benzylester formation is obtained in the presence of benzylamine upon selective alcohol activation by the NHC. High level DFT calculations reveal that alcohol activation occurs by strong H-bond formation between the NHC and the alcohol thereby increasing the nucleophilicity of the alcohol. For oxidatively generated acylazolium ions under NHC catalysis, the carbene has a dual role (cooperative catalysis): (a) the NHC is used for generation of the acylazolium ion and (b) the NHC is used for activation of the alcohol in the subsequent acylation step. NHC-catalyzed selective acylation of benzyl alcohol in the presence of benzylamine can also be achieved with trifluoroethyl and hexafluoroisopropylesters as acylation reagents. Moreover, an enol acetate also shows high O-selectivity as a chemoselective acetylation reagent. Kinetic and mechanistic studies are provided and some examples of the chemoselective acylation of amino alcohols are presented.
Co-reporter:Tobias Risthaus and Stefan Grimme
Journal of Chemical Theory and Computation 2013 Volume 9(Issue 3) pp:1580-1591
Publication Date(Web):January 29, 2013
DOI:10.1021/ct301081n
A new test set (S12L) containing 12 supramolecular noncovalently bound complexes is presented and used to evaluate seven different methods to account for dispersion in DFT (DFT-D3, DFT-D2, DFT-NL, XDM, dDsC, TS-vdW, M06-L) at different basis set levels against experimental, back-corrected reference energies. This allows conclusions about the performance of each method in an explorative research setting on “real-life” problems. Most DFT methods show satisfactory performance but, due to the largeness of the complexes, almost always require an explicit correction for the nonadditive Axilrod–Teller–Muto three-body dispersion interaction to get accurate results. The necessity of using a method capable of accounting for dispersion is clearly demonstrated in that the two-body dispersion contributions are on the order of 20–150% of the total interaction energy. MP2 and some variants thereof are shown to be insufficient for this while a few tested D3-corrected semiempirical MO methods perform reasonably well. Overall, we suggest the use of this benchmark set as a “sanity check” against overfitting to too small molecular cases.
Co-reporter:Waldemar Hujo and Stefan Grimme
Journal of Chemical Theory and Computation 2013 Volume 9(Issue 1) pp:308-315
Publication Date(Web):October 26, 2012
DOI:10.1021/ct300813c
The nonlocal, electron density dependent dispersion correction of Vydrov and Van Voorhis (Vydrov, O. A.; Van Voorhis, T. J. Chem. Phys.2010, 133, 244103), termed VV10 or DFT-NL, has been implemented for structural optimizations of molecules. It is tested in combination with the four (hybrid)GGA density functionals TPSS, TPSS0, B3LYP, and revPBE38 for inter- and intramolecular noncovalent interactions (NCI) and compared to results from atom-pairwise dispersion corrected DFT-D3. The methods are applied to a wide range of different problems, namely the S22 and S66 test sets, large transition metal complexes, water hexamer clusters, hexahelicene, and four other difficult cases of intramolecular NCI. Critical interatomic distances are computed remarkably accurately by both dispersion corrections compared to theoretical or experimental reference data and inter- and intramolecular interactions are treated on equal footing. The methods can be recommended as reliable and robust tools for geometry optimizations of large systems in which long-range dispersion forces are crucial.
Co-reporter:Stefan Grimme and Marc Steinmetz
Physical Chemistry Chemical Physics 2013 vol. 15(Issue 38) pp:16031-16042
Publication Date(Web):11 Jul 2013
DOI:10.1039/C3CP52293H
A benchmark set of 25 rotational constants measured in the gas phase for nine molecules (termed ROT25) was compiled from available experimental data. The medium-sized molecules with 18–35 atoms cover common (bio)organic structure motifs including hydrogen bonding and flexible side chains. They were each considered in a single conformation. The experimental B0 values were back-corrected to reference equilibrium rotational constants (Be) by computation of the vibrational corrections ΔBvib. Various density functional theory (DFT) methods and Hartree–Fock with and without dispersion corrections as well as MP2 type methods and semi-empirical quantum chemical approaches are investigated. The ROT25 benchmark tests their ability to describe covalent bond lengths, longer inter-atomic distances, and the relative orientation of functional groups (intramolecular non-covalent interactions). In general, dispersion corrections to DFT and HF increase Be values (shrink molecular size) significantly by about 0.5–1.5% thereby in general improving agreement with the reference data. Regarding DFT methods, the overall accuracy of the optimized structures roughly follows the ‘Jacobs ladder’ classification scheme, i.e., it decreases in the series double-hybrid > (meta)hybrid > (meta)GGA > LDA. With B2PLYP-D3, SCS-MP2, B3LYP-D3/NL, or PW6B95-D3 methods and extended QZVP (def2-TZVP) AO basis sets, Be values, accurate to about 0.3–0.6 (0.5–1)% on average, can be computed routinely. The accuracy of B2PLYP-D3/QZVP with a mean deviation of only 3 MHz and a standard deviation of 0.24% is exceptional and we recommend this method when highly accurate structures are required or for problematic conformer assignments. The correlation effects for three inter-atomic distance regimes (covalent, medium-range, long) and the performance of minimal basis set (semi-empirical) methods are discussed.
Co-reporter:Jan Gerit Brandenburg, Maristella Alessio, Bartolomeo Civalleri, Michael F. Peintinger, Thomas Bredow, and Stefan Grimme
The Journal of Physical Chemistry A 2013 Volume 117(Issue 38) pp:9282-9292
Publication Date(Web):August 15, 2013
DOI:10.1021/jp406658y
We extend the previously developed geometrical correction for the inter- and intramolecular basis set superposition error (gCP) to periodic density functional theory (DFT) calculations. We report gCP results compared to those from the standard Boys–Bernardi counterpoise correction scheme and large basis set calculations. The applicability of the method to molecular crystals as the main target is tested for the benchmark set X23. It consists of 23 noncovalently bound crystals as introduced by Johnson et al. ( J. Chem. Phys. 2012, 137, 054103) and refined by Tkatchenko et al. ( J. Chem. Phys. 2013, 139, 024705). In order to accurately describe long-range electron correlation effects, we use the standard atom-pairwise dispersion correction scheme DFT-D3. We show that a combination of DFT energies with small atom-centered basis sets, the D3 dispersion correction, and the gCP correction can accurately describe van der Waals and hydrogen-bonded crystals. Mean absolute deviations of the X23 sublimation energies can be reduced by more than 70% and 80% for the standard functionals PBE and B3LYP, respectively, to small residual mean absolute deviations of about 2 kcal/mol (corresponding to 13% of the average sublimation energy). As a further test, we compute the interlayer interaction of graphite for varying distances and obtain a good equilibrium distance and interaction energy of 6.75 Å and −43.0 meV/atom at the PBE-D3-gCP/SVP level. We fit the gCP scheme for a recently developed pob-TZVP solid-state basis set and obtain reasonable results for the X23 benchmark set and the potential energy curve for water adsorption on a nickel (110) surface.
Co-reporter:Jonas Moellmann and Stefan Grimme
Organometallics 2013 Volume 32(Issue 14) pp:3784-3787
Publication Date(Web):July 3, 2013
DOI:10.1021/om400386x
The chemically relevant intramolecular bond distance between a η3-allylic system and the metal center in a ruthenium complex has been reinvestigated by state-of-the-art dispersion-corrected DFT calculations. Previous works revealed significant deviations between the experimental X-ray crystal structure and results of molecular DFT gas-phase calculations. It was stated that this complex represents a general failure of DFT for transition-metal complex geometries. Our work shows that periodic DFT-D3 calculations for the crystal excellently reproduce all experimental data. The previously found deviations are attributed to crystal packing, the counterions, and improperly accounted dispersion interactions in the theoretical treatment.
Co-reporter:Jens Antony, Bassam Alameddine, Titus A. Jenny, and Stefan Grimme
The Journal of Physical Chemistry A 2013 Volume 117(Issue 3) pp:616-625
Publication Date(Web):December 26, 2012
DOI:10.1021/jp3075207
Stacked dimers of four polycondensed aromatic hydrocarbons, with structures varying from high to reduced symmetries, have been calculated with dispersion-corrected density functional theory. The configurations of the stacked dimers are readily classified by two in-plane displacements and a relative rotation. The potential energy surface in these three coordinates was calculated with rigid monomers and appears to be slightly flat. Full geometry optimization was performed for selected low-energy structures, resulting in an energy ranking of a series of conformations whose geometries were characterized in considerable detail. The dissociation energy values reveal a clear preference for the symmetrical disk-shaped and triangular structures to dimerize into two in-plane-displaced arrangements, whereas the less symmetrical trapezoidal structures show a tendency to stack in displaced antiparallel over parallel arrangements. According to methodical checks, the key computational results, namely, the shape of the potential energy surface and the geometrical structures and energy ranking of dimer conformations, are essentially insensitive to computational assumptions such as the atomic orbital basis set and density functional chosen. This is shown in particular for the basis set superposition error, which, for the selected level of theory [B97-D3(BJ)/TZV(d,p)] was estimated by the counterpoise correction procedure to be in the narrow range between 7% and 8% of the uncorrected dissociation energies.
Co-reporter:Marc Steinmetz
ChemistryOpen 2013 Volume 2( Issue 3) pp:115-124
Publication Date(Web):
DOI:10.1002/open.201300012
Abstract
The performance of 23 density functionals, including one LDA, four GGAs, three meta-GGAs, three hybrid GGAs, eight hybrid meta-GGAs, and ten double-hybrid functionals, was investigated for the computation of activation energies of various covalent main-group single bonds by four catalysts: Pd, PdCl−, PdCl2, and Ni (all in the singlet state). A reactant complex, the barrier, and reaction energy were considered, leading to 164 energy data points for statistical analysis. Extended Gaussian AO basis sets were used in all calculations. The best functional for the complete benchmark set relative to estimated CCSD(T)/CBS reference data is PBE0-D3, with an MAD value of 1.1 kcal mol−1 followed by PW6B95-D3, the double hybrid PWPB95-D3, and B3LYP-D3 (1.9 kcal mol−1 each). The other tested hybrid meta-GGAs perform less well (M06-HF: 7.0 kcal mol−1; M06-2X: 6.3 kcal mol−1; M06: 4.9 kcal mol−1) for the investigated reactions. In the Ni case, some double hybrids show larger errors due to partial breakdown of the perturbative treatment for the correlation energy in cases with difficult electronic structures (partial multi-reference character). Only double hybrids either with very low amounts of perturbative correlation (e.g., PBE0-DH) or that use the opposite-spin correlation component only (e.g., PWPB95) seem to be more robust. We also investigated the effect of the D3 dispersion correction. While the barriers are not affected by this correction, significant and mostly positive results were observed for reaction energies. Furthermore, six very recently proposed double-hybrid functionals were analyzed regarding the influence of the amount of Fock exchange as well as the type of perturbative correlation treatment. According to these results, double hybrids with <50–60 % of exact exchange and ∼30 % perturbative correlation perform best.
Co-reporter:Jan Gerit Brenburg;Dr. Stefan Grimme;Dr. Peter G. Jones;Dr. Georgios Markopoulos;Dr. Henning Hopf; Dr. Micha&x142; K. Cyranski;Dr. Dietmar Kuck
Chemistry - A European Journal 2013 Volume 19( Issue 30) pp:9930-9938
Publication Date(Web):
DOI:10.1002/chem.201300761
Abstract
A combined X-ray diffraction and theoretical study of the solid-state molecular and crystal structures of tribenzotriquinacene (TBTQ, 2) and its centro-methyl derivative (3) is presented. The molecular structure of the parent hydrocarbon displays C3v symmetry and the three indane wings adopt mutually orthogonal orientations, similar to the case in its previously reported methyl derivative (3). Also similarly to the latter structure, the bowl-shaped molecules of compound 2 form infinite molecular stacks with perfectly axial, face-to-back (convex–concave) packing and with parallel and unidirectional orientation of the stacks. The experimentally determined intra-stack molecular distance is 4.75 Å for compound 2 and 5.95 Å for compound 3. Whereas the molecules of compound 2 show a slight alternating rotation (±6°) about the common axis of each stack, those of compound 3 show perfect translational symmetry within the stacks. We used dispersion-corrected density functional theory to compute the crystal structures of tribenzotriquinacenes 2 and 3. The London dispersion correction was crucial for obtaining an accurate description of the crystallization of both analyzed systems and the calculated results agreed excellently with the experimental measurements. We also obtained reasonable sublimation energies for both compounds. In addition, the geometries and dimerization energies of oligomeric stacks of compound 2 were computed and showed smooth convergence to the properties of the infinite polymeric stack.
Co-reporter:Dr. Stephan Ehrlich;Dr. Holger F. Bettinger;Dr. Stefan Grimme
Angewandte Chemie 2013 Volume 125( Issue 41) pp:11092-11096
Publication Date(Web):
DOI:10.1002/ange.201304674
Co-reporter:Gabi Ohlendorf;Dr. Christian W. Mahler;Dr. Stefan-S. Jester;Dr. Gregor Schnakenburg;Dr. Stefan Grimme;Dr. Sigurd Höger
Angewandte Chemie 2013 Volume 125( Issue 46) pp:12308-12312
Publication Date(Web):
DOI:10.1002/ange.201306299
Co-reporter:Dr. Stefan Grimme
Angewandte Chemie International Edition 2013 Volume 52( Issue 24) pp:6306-6312
Publication Date(Web):
DOI:10.1002/anie.201300158
Co-reporter:Dr. Stephan Ehrlich;Dr. Holger F. Bettinger;Dr. Stefan Grimme
Angewandte Chemie International Edition 2013 Volume 52( Issue 41) pp:10892-10895
Publication Date(Web):
DOI:10.1002/anie.201304674
Co-reporter:Gabi Ohlendorf;Dr. Christian W. Mahler;Dr. Stefan-S. Jester;Dr. Gregor Schnakenburg;Dr. Stefan Grimme;Dr. Sigurd Höger
Angewandte Chemie International Edition 2013 Volume 52( Issue 46) pp:12086-12090
Publication Date(Web):
DOI:10.1002/anie.201306299
Co-reporter:Dr. Stefan Grimme
Angewandte Chemie 2013 Volume 125( Issue 24) pp:6426-6433
Publication Date(Web):
DOI:10.1002/ange.201300158
Co-reporter:Thomas Wiegand ; Hellmut Eckert ; Olga Ekkert ; Roland Fröhlich ; Gerald Kehr ; Gerhard Erker ≠
Journal of the American Chemical Society 2012 Volume 134(Issue 9) pp:4236-4249
Publication Date(Web):January 20, 2012
DOI:10.1021/ja210160k
Covalent bonding interactions between the Lewis acid and Lewis base functionalities have been probed in a series of “frustrated Lewis pairs” (FLPs) (mainly substituted vinylene linked intramolecular phosphane–borane adducts), using solid-state nuclear magnetic resonance techniques and accompanying DFT calculations. Both the 11B NMR isotropic chemical shifts and nuclear electric quadrupolar coupling parameters turn out to be extremely sensitive experimental probes for such interactions, revealing linear correlations with boron–phosphorus internuclear distances. The principal component Vzz of the 11B electric field gradient tensor is tilted slightly away (∼20°) from the boron–phosphorus internuclear vector, leading to an improved understanding of the remarkable reactivity of the FLPs. Complementary 31P{1H}-CPMAS experiments reveal significant 31P–11B scalar spin–spin interactions (1J ≈ 50 Hz), evidencing covalent bonding interactions between the reaction centers. Finally, 11B{31P} rotational echo double resonance (REDOR) experiments show systematic deviations from calculated curves based on the internuclear distances from X-ray crystallography. These deviations suggest non-zero contributions from anisotropic indirect spin–spin (J anisotropy) interactions, thereby offering additional evidence for covalent bonding.
Co-reporter:Muhammad Sajid ; Annika Stute ; Allan Jay P. Cardenas ; Brooks J. Culotta ; Johannes A. M. Hepperle ; Timothy H. Warren ; Birgitta Schirmer ; Stefan Grimme ; Armido Studer ; Constantin G. Daniliuc ; Roland Fröhlich ; Jeffrey L. Petersen ; Gerald Kehr ;Gerhard Erker
Journal of the American Chemical Society 2012 Volume 134(Issue 24) pp:10156-10168
Publication Date(Web):May 1, 2012
DOI:10.1021/ja302652a
The intramolecular cyclohexylene-bridged P/B frustrated Lewis pair [Mes2P—C6H10—B(C6F5)2] 1b reacts rapidly with NO to give the persistent FLP-NO aminoxyl radical 2b formed by P/B addition to the nitrogen atom of NO. This species was fully characterized by X-ray diffraction, EPR and UV/vis spectroscopies, C,H,N elemental analysis, and DFT calculations. The reactive oxygen-centered radical 2b undergoes a H-atom abstraction (HAA) reaction with 1,4-cyclohexadiene to give the diamagnetic FLP-NOH product 3b. FLP-NO 2b reacts with toluene at 70 °C in an HAA/radical capture sequence to give a 1:1 mixture of FLP-NOH 3b and FLP-NO—CH2Ph 4b, both characterized by X-ray diffraction. Structurally related FLPs [Mes2P—CHR1—CHR2—B(C6F5)2] 1c, 1d, and 1e react analogously with NO to give the respective persistent FLP-NO radicals 2c, 2d, and 2e, respectively, which show similar HAA and O-functionalization reactions. The FLP-NO—CHMePh 6b derived from 1-bromoethylbenzene undergoes NO—C bond cleavage at 120 °C with an activation energy of Ea = 35(2) kcal/mol. Species 6b induces the controlled nitroxide-mediated radical polymerization (NMP) of styrene at 130 °C to give polystyrene with a polydispersity index of 1.3. The FLP-NO systems represent a new family of aminoxyl radicals that are easily available by N,N-cycloaddition of C2-bridged intramolecular P/B frustrated Lewis pairs to nitric oxide.
Co-reporter:Stefan Grimme, Waldemar Hujo and Barbara Kirchner
Physical Chemistry Chemical Physics 2012 vol. 14(Issue 14) pp:4875-4883
Publication Date(Web):29 Feb 2012
DOI:10.1039/C2CP24096C
Potential energy curves for the dissociation of cation–anion associates representing the building units of ionic liquids have been computed with dispersion corrected DFT methods. Non-local van der Waals density functionals (DFT-NL) for the first time as well as an atom pair-wise correction method (DFT-D3) have been tested. Reference data have been computed at the extrapolated MP2/CBS and estimated CCSD(T)/CBS levels of theory. The investigated systems are combined from two cations (1-butyl-3-methylimidazolium and tributyl(methyl)posphonium) and three anions (chloride, dicyanamide, acetate). We find substantial stabilization from London dispersion energy near equilibrium of 5–7 kcal mol−1 (about 5–6% of the interaction energy). Equilibrium distances are shortened by 0.03–0.09 Å and fundamental (inter-fragment) vibrational frequencies (which are in the range 140–180 cm−1) are increased by typically 10 cm−1 when dispersion corrections are made. The dispersion-corrected hybrid functional potentials are in general in excellent agreement with the corresponding CCSD(T) reference data (typical deviations of about 1–2%). The DFT-D3 method performs unexpectedly well presumably because of cancellation of errors between the dispersion coefficients of the cations and anions. Due to self-interaction error, semi-local density functionals exhibit severe SCF convergence problems, and provide artificial charge-transfer and inaccurate interaction energies for larger inter-fragment distances. Although these problems may be alleviated in condensed phase simulations by effective Coulomb screening, only dispersion-corrected hybrid functionals with larger amounts of Fock-exchange can in general be recommended for such ionic systems.
Co-reporter:Christian Mück-Lichtenfeld and Stefan Grimme
Dalton Transactions 2012 vol. 41(Issue 30) pp:9111-9118
Publication Date(Web):03 May 2012
DOI:10.1039/C2DT30562C
The energy profiles of the activation reaction of small molecules (H2, Br2 and CO2) with boron/phosphorus frustrated Lewis pairs (FLPs) have been calculated with dispersion corrected DFT (TPSS-D3). We have investigated the cooperative nature of the reactions by analyzing interaction energies in the ternary system and for reactant pairs. The non-additive contributions to the total interaction energy add to the driving force of the activation reaction, even at early stages of the process. We propose the isosurface representation of the many-body deformation density Δρmb as a qualitative tool to visualize cooperative, non-additive effects in complex chemical systems.
Co-reporter:S. Ehrlich, S. Grimme
Computational and Theoretical Chemistry 2012 Volume 999() pp:152-153
Publication Date(Web):1 November 2012
DOI:10.1016/j.comptc.2012.08.028
Modern dispersion corrected density functional theory (DFT-D3) can reproduce accurate wave function based results (est. CCSD (T)/CBS) for the non-covalent interactions of different conformers of the thioformyl chloride dimer. The failure of B2-PLYP-D reported in the original publication can be attributed to an insufficient description of third-row elements in the older versions of DFT-D, which also holds to some degree for the electronic part of B97-D. This, as well as the flatness of the potential energy surface of the studied complexes, is the main reason for the reported failure of the two methods.Graphical abstractHighlights► Failure of B2-PLYP-D on the test set can be attributed to use of DFT-D2. ► Use of DFT-D3 improves results for the test set. ► Flat potential energy surfaces should be handled with care.
Co-reporter:Wenliang Li, Stefan Grimme, Helge Krieg, Jonas Möllmann, and Jingping Zhang
The Journal of Physical Chemistry C 2012 Volume 116(Issue 16) pp:8865-8871
Publication Date(Web):April 5, 2012
DOI:10.1021/jp2112632
Microporous organic molecular crystals (MOMCs) are materials composed of discrete organic molecules interacting noncovalently. The gas uptake of CO2, CH4, N2, and Xe in one of the most widely investigated MOMCs, trispiro(benzodioxole[2′.2:2″.4:2‴.6]1,3,5,2,4,6-triazatriphosphinine) (1), was computed by grand canonical Monte Carlo (GCMC) simulations based on our lately developed force field method vdW3, which is fitted to accurate ab initio potentials (double hybrid functional B2PLYP with a D3 dispersion correction using def2-TZVPP basis sets). The B2PLYP-D3 results compare very well with CCSD(T)/CBS interaction energies for benzene–gas model complexes. Our multiscale modeling approach to accurate interaction potentials is found to be essential in order to obtain reasonable adsorption isotherms and heats of adsorption. The good agreement between the simulation results and experimental data in all cases gives us the opportunity to design novel complex materials for gas adsorption based solely on first-principles calculations.
Co-reporter:Marc Steinmetz;Kirika Ueda;Dr. Stefan Grimme;Dr. Junichiro Yamaguchi;Dr. Sylvia Kirchberg;Dr. Kenichiro Itami;Dr. Armido Studer
Chemistry – An Asian Journal 2012 Volume 7( Issue 6) pp:1256-1260
Publication Date(Web):
DOI:10.1002/asia.201101011
Abstract
Direct CH phenylation of 2-ethylthiophene and 2-chlorothiophene with PhPdI(bipy) complex to form either the corresponding 4-phenyl or 5-phenylthiophene derivative is studied under stoichiometric conditions using various Lewis acids as additives. It is shown that reactions occur via the corresponding cationic Pd complex (PhPdbipy+) and that the counteranion determines the regioselectivity. High-level DFT calculations reveal that CC bond formation occurs via a carbopalladation pathway and not via electrophilic palladation. These calculations give some indications regarding the regioselectivity of the thiophene arylation.
Co-reporter:Lutz Greb;Dr. Pascual Oña-Burgos;Birgitta Schirmer;Dr. Stefan Grimme;Dr. Douglas W. Stephan;Dr. Jan Paradies
Angewandte Chemie 2012 Volume 124( Issue 40) pp:10311-10315
Publication Date(Web):
DOI:10.1002/ange.201204007
Co-reporter:Holger Kruse, Lars Goerigk, and Stefan Grimme
The Journal of Organic Chemistry 2012 Volume 77(Issue 23) pp:10824-10834
Publication Date(Web):November 15, 2012
DOI:10.1021/jo302156p
We analyze the error compensations that are responsible for the relatively good performance of the popular B3LYP/6-31G* model chemistry for molecular thermochemistry. We present the B3LYP-gCP-D3/6-31G* scheme, which corrects for missing London dispersion and basis set superposition error (BSSE) in a physically sound manner. Benchmark results for the general main group thermochemistry, kinetics, and noncovalent interactions set (GMTKN30) are presented. A detailed look is cast on organic reactions of several arenes with C60, Diels–Alder reactions, and barriers to [4 + 3] cycloadditions. We demonstrate the practical advantages of the new B3LYP-gCP-D3/6-31G* scheme and show its higher robustness over standard B3LYP/6-31G*. B3LYP-gCP-D3/6-31G* is meant to fully substitute standard B3LYP/6-31G* calculations in the same black-box sense at essentially no increase in computational cost. The energy corrections are made available by a Web service (http://www.thch.uni-bonn.de/tc/gcpd3) and by freely available software.
Co-reporter: Dr. Stefan Grimme
Chemistry - A European Journal 2012 Volume 18( Issue 32) pp:9955-9964
Publication Date(Web):
DOI:10.1002/chem.201200497
Abstract
The equilibrium association free enthalpies ΔGa for typical supramolecular complexes in solution are calculated by ab initio quantum chemical methods. Ten neutral and three positively charged complexes with experimental ΔGa values in the range 0 to −21 kcal mol−1 (on average −6 kcal mol−1) are investigated. The theoretical approach employs a (nondynamic) single-structure model, but computes the various energy terms accurately without any special empirical adjustments. Dispersion corrected density functional theory (DFT-D3) with extended basis sets (triple-ζ and quadruple-ζ quality) is used to determine structures and gas-phase interaction energies (ΔE), the COSMO-RS continuum solvation model (based on DFT data) provides solvation free enthalpies and the remaining ro-vibrational enthalpic/entropic contributions are obtained from harmonic frequency calculations. Low-lying vibrational modes are treated by a free-rotor approximation. The accurate account of London dispersion interactions is mandatory with contributions in the range −5 to −60 kcal mol−1 (up to 200 % of ΔE). Inclusion of three-body dispersion effects improves the results considerably. A semilocal (TPSS) and a hybrid density functional (PW6B95) have been tested. Although the ΔGa values result as a sum of individually large terms with opposite sign (ΔE vs. solvation and entropy change), the approach provides unprecedented accuracy for ΔGa values with errors of only 2 kcal mol−1 on average. Relative affinities for different guests inside the same host are always obtained correctly. The procedure is suggested as a predictive tool in supramolecular chemistry and can be applied routinely to semirigid systems with 300–400 atoms. The various contributions to binding and enthalpy–entropy compensations are discussed.
Co-reporter: Stefan Grimme
ChemPhysChem 2012 Volume 13( Issue 6) pp:1407-1409
Publication Date(Web):
DOI:10.1002/cphc.201200094
No abstract is available for this article.
Co-reporter:Lutz Greb;Dr. Pascual Oña-Burgos;Birgitta Schirmer;Dr. Stefan Grimme;Dr. Douglas W. Stephan;Dr. Jan Paradies
Angewandte Chemie International Edition 2012 Volume 51( Issue 40) pp:10164-10168
Publication Date(Web):
DOI:10.1002/anie.201204007
Co-reporter:Waldemar Hujo and Stefan Grimme
Journal of Chemical Theory and Computation 2011 Volume 7(Issue 12) pp:3866-3871
Publication Date(Web):October 25, 2011
DOI:10.1021/ct200644w
The nonlocal van der Waals density functional VV10 (Vydrov, O. A.; Van Voorhis, T. J. Chem. Phys.2010, 133, 244103) is tested for the thermochemical properties of 1200+ atoms and molecules in the GMTKN30 database in order to assess its global accuracy. Five GGA and hybrid functionals in unmodified form are augmented by the nonlocal (NL) part of the VV10 functional (one parameter adjusted). The addition of the NL dispersion energy definitely improves the results of all tested functionals. On the basis of little empiricism and basic physical insight, DFT-NL can be recommended as a fully electronic, robust electronic structure method.
Co-reporter:Dr. Lars Goerigk;Holger Kruse; Dr. Stefan Grimme
ChemPhysChem 2011 Volume 12( Issue 17) pp:3421-3433
Publication Date(Web):
DOI:10.1002/cphc.201100826
Abstract
Dispersion-corrected density functional theory is assessed on the new S66 and S66x8 benchmark sets for non-covalent interactions. In total, 17 different density functionals are evaluated. Two flavors of our latest additive London-dispersion correction DFT-D3 and DFT-D3(BJ), which differ in their short-range damping functions, are tested. In general, dispersion corrections are again shown to be crucial to obtain reliable non-covalent interaction energies and equilibrium distances. The corrections strongly diminish the performance differences between the functionals, and in summary most dispersion-corrected methods can be recommended. DFT-D3 and DFT-D3(BJ) also yield similar results but for most functionals and intermolecular distances, the rational Becke–Johnson scheme performs slightly better. Particularly, the statistical analysis for S66x8, which covers also non-equilibrium complex geometries, shows that the Minnesota class of functionals is also improved by the D3 scheme. The best methods on the (meta-)GGA or hybrid- (meta-)GGA level are B97-D3, BLYP-D3(BJ), PW6B95-D3, MPW1B95-D3 and LC-ωPBE-D3. Double-hybrid functionals are the most accurate and robust methods, and in particular PWPB95-D3 and B2-PLYP-D3(BJ) can be recommended. The best DFT-D3 and DFT-D3(BJ) approaches are competitive to specially adapted perturbation methods and clearly outperform standard MP2. Comparisons between S66, S22 and parts of the GMTKN30 database show that the S66 set provides statistically well-behaved data and can serve as a valuable tool for, for example, fitting purposes or cross-validation of other benchmark databases.
Co-reporter:Stephan Ehrlich;Jonas Moellmann;Dr. Werner Reckien; Dr. Thomas Bredow; Dr. Stefan Grimme
ChemPhysChem 2011 Volume 12( Issue 17) pp:3414-3420
Publication Date(Web):
DOI:10.1002/cphc.201100521
Abstract
Dispersion-corrected density functional theory calculations (DFT-D3) were performed for the adsorption of CO on MgO and C2H2 on NaCl surfaces. An extension of our non-empirical scheme for the computation of atom-in-molecules dispersion coefficients is proposed. It is based on electrostatically embedded M4X4 (MNa, Mg) clusters that are used in TDDFT calculations of dynamic dipole polarizabilities. We find that the dispersion coefficients for bulk NaCl and MgO are reduced by factors of about 100 and 35 for Na and Mg, respectively, compared to the values of the free atoms. These are used in periodic DFT calculations with the revPBE semi-local density functional. As demonstrated by calculations of adsorption potential energy curves, the new C6 coefficients lead to much more accurate energies (Eads) and molecule–surface distances than with previous DFT-D schemes. For NaCl/C2H2 we obtained at the revPBE-D3(BJ) level a value of Eads=−7.4 kcal mol−1 in good agreement with experimental data (−5.7 to −7.1 kcal mol−1). Dispersion-uncorrected DFT yields an unbound surface state. For the MgO/CO system, the computed revPBE-D3(BJ) value of Eads=−4.1 kcal mol−1 is also in reasonable agreement with experimental results (−3.0 kcal mol−1) when thermal corrections are taken into account. Our new dispersion correction also improves computed lattice constants of the bulk systems significantly compared to plain DFT or previous DFT-D results. The extended DFT-D3 scheme also provides accurate non-covalent interactions for ionic systems without empirical adjustments and is suggested as a general tool in surface science.
Co-reporter:Dr. Stefan Grimme;Dr. Peter R. Schreiner
Angewandte Chemie 2011 Volume 123( Issue 52) pp:12849-12853
Publication Date(Web):
DOI:10.1002/ange.201103615
Co-reporter: Dr. Victor S. Batista; Dr. Stefan Grimme; Dr. Markus Reiher
ChemPhysChem 2011 Volume 12( Issue 17) pp:3043-3044
Publication Date(Web):
DOI:10.1002/cphc.201100862
No abstract is available for this article.
Co-reporter:Dr. Stefan Grimme;Dr. Peter R. Schreiner
Angewandte Chemie International Edition 2011 Volume 50( Issue 52) pp:12639-12642
Publication Date(Web):
DOI:10.1002/anie.201103615
Co-reporter:Rebecca Sure, Andreas Hansen, Peter Schwerdtfeger and Stefan Grimme
Physical Chemistry Chemical Physics 2017 - vol. 19(Issue 22) pp:NaN14305-14305
Publication Date(Web):2017/05/18
DOI:10.1039/C7CP00735C
We investigate all 1812 isomers of the fullerene C60 consistently with high-accuracy quantum chemistry methods. The isomers are optimized at the PBE-D3/def2-TZVP level of theory and their relative energies are obtained at the affordable and accurate hybrid-DFT level PW6B95-D3ATM/def2-QZVP. For the first time reliable values for the relative energy distribution among the 1812 isomers (maximum value of 549.1 kcal mol−1, average value of 189.8 ± 46.8 kcal mol−1, 1267 isomers in an energy window of 150–250 kcal mol−1) are given. The quality of the DFT energies is verified by comparison with highly accurate DLPNO-CCSD(T)/CBS* results for ten selected C60 isomers, and the DFT results are further used to benchmark several semiempirical methods. Our findings yield methodological insight into future multi-level modelling of larger carbon nano-structures. We correlate the best relative energies of the isomers to a number of topological indices, electronic properties, and geometrical measures in order to rationalize the isomeric stability aside from the common isolated pentagon rule minimizing the number of pentagon fusions. The information from the best qualitative measures can be condensed to the statement, that a small pentagon signature P1, a large volume, and a more spherical cage lead to a relatively stable isomer. In addition, the best semiempirical method was applied to all 31924 isomers of C80.
Co-reporter:Vilhjálmur Ásgeirsson, Christoph A. Bauer and Stefan Grimme
Chemical Science (2010-Present) 2017 - vol. 8(Issue 7) pp:NaN4895-4895
Publication Date(Web):2017/05/05
DOI:10.1039/C7SC00601B
We introduce a fully stand-alone version of the Quantum Chemistry Electron Ionization Mass Spectra (QCEIMS) program [S. Grimme, Angew. Chem. Int. Ed., 2013, 52, 6306] allowing efficient simulations for molecules composed of elements with atomic numbers up to Z = 86. The recently developed extended tight-binding semi-empirical method GFN-xTB has been combined with QCEIMS, thereby eliminating dependencies on third-party electronic structure software. Furthermore, for reasonable calculations of ionization potentials, as required by the method, a second tight-binding variant, IPEA-xTB, is introduced here. This novel combination of methods allows the automatic, fast and reasonably accurate computation of electron ionization mass spectra for structurally different molecules across the periodic table. In order to validate and inspect the transferability of the method, we perform large-scale simulations for some representative organic, organometallic, and main-group inorganic systems. Theoretical spectra for 23 molecules are compared directly to experimental data taken from standard databases. For the first time, realistic quantum chemistry based EI-MS for organometallic systems like ferrocene or copper(II)acetylacetonate are presented. Compared to previously used semiempirical methods, GFN-xTB is faster, more robust, and yields overall higher quality spectra. The partially analysed theoretical reaction and fragmentation mechanisms are chemically reasonable and reveal in unprecedented detail the extreme complexity of high energy gas phase ion chemistry including complicated rearrangement reactions prior to dissociation.
Co-reporter:Christian Honacker, Zheng-Wang Qu, Jens Tannert, Marcus Layh, Alexander Hepp, Stefan Grimme and Werner Uhl
Dalton Transactions 2016 - vol. 45(Issue 14) pp:NaN6174-6174
Publication Date(Web):2015/11/19
DOI:10.1039/C5DT03918E
Treatment of alkynyl–arylchlorogermanes ArylnGe(Cl)(CC–tBu)3−n (n = 1, 2) with HMtBu2 (M = Al, Ga) yielded mixed Al or Ga alkenyl–alkynylchlorogermanes via hydrometallation reactions. Intramolecular interactions between the Lewis-basic Cl atoms and the Lewis-acidic Al or Ga atoms afforded MCGeCl heterocycles. The endocyclic M–Cl distances were significantly lengthened compared to the starting compounds and indicated Ge–Cl bond activation. Dual hydrometallation succeeded only with HGatBu2. One Ga atom of the product was involved in a Ga–Cl bond, while the second one had an interaction to a C–H bond of a phenyl group. In two cases treatment of chlorogermanes with two equivalents of HAltBu2 resulted in hydroalumination of one alkynyl group and formation of unprecedented Ge–H functionalized germanes, Aryl–Ge(H)(CC–tBu)[C(AltBu2)C(H)–tBu] (Aryl = mesityl, triisopropylphenyl). The Al atoms of these compounds interacted with the α-C atoms of the alkynyl groups. Ph(Cl)Ge(CC–tBu)[C(AltBu2}C(H)–tBu] reacted in an unusual Cl/tBu exchange to yield the tert-butylgermane Ph(tBu)Ge(CC–tBu)[C{Al(tBu)(Cl)}C(H)–tBu]. Quantum chemical calculations suggested the formation of a germyl cation as a transient intermediate.
Co-reporter:Christoph Alexander Bauer and Stefan Grimme
Organic & Biomolecular Chemistry 2014 - vol. 12(Issue 43) pp:NaN8744-8744
Publication Date(Web):2014/09/08
DOI:10.1039/C4OB01668H
This study presents a showcase for the novel Quantum Chemistry Electron Ionization Mass Spectrometry (QCEIMS) method on five FDA-approved drugs. The method allows a first-principles electronic structure-based prediction of EI mass spectra in principle for any molecule. The systems in this case study are organic substances of nominal masses between 404 and 853 atomic mass units and cover a wide range of functional groups and organic molecular structure motifs. The results demonstrate the widespread applicability of the QCEIMS method for the unbiased computation of EI mass spectra even for larger molecules. Its strengths compared to standard (static) or database driven approaches in such cases are highlighted. Weak points regarding the required computation times or the approximate character of the employed QC methods are also discussed. We propose QCEIMS as a viable and robust way of predicting EI mass spectra for sizeable organic molecules relevant to medicinal and pharmaceutical chemistry.
Co-reporter:Stefan Grimme, Waldemar Hujo and Barbara Kirchner
Physical Chemistry Chemical Physics 2012 - vol. 14(Issue 14) pp:NaN4883-4883
Publication Date(Web):2012/02/29
DOI:10.1039/C2CP24096C
Potential energy curves for the dissociation of cation–anion associates representing the building units of ionic liquids have been computed with dispersion corrected DFT methods. Non-local van der Waals density functionals (DFT-NL) for the first time as well as an atom pair-wise correction method (DFT-D3) have been tested. Reference data have been computed at the extrapolated MP2/CBS and estimated CCSD(T)/CBS levels of theory. The investigated systems are combined from two cations (1-butyl-3-methylimidazolium and tributyl(methyl)posphonium) and three anions (chloride, dicyanamide, acetate). We find substantial stabilization from London dispersion energy near equilibrium of 5–7 kcal mol−1 (about 5–6% of the interaction energy). Equilibrium distances are shortened by 0.03–0.09 Å and fundamental (inter-fragment) vibrational frequencies (which are in the range 140–180 cm−1) are increased by typically 10 cm−1 when dispersion corrections are made. The dispersion-corrected hybrid functional potentials are in general in excellent agreement with the corresponding CCSD(T) reference data (typical deviations of about 1–2%). The DFT-D3 method performs unexpectedly well presumably because of cancellation of errors between the dispersion coefficients of the cations and anions. Due to self-interaction error, semi-local density functionals exhibit severe SCF convergence problems, and provide artificial charge-transfer and inaccurate interaction energies for larger inter-fragment distances. Although these problems may be alleviated in condensed phase simulations by effective Coulomb screening, only dispersion-corrected hybrid functionals with larger amounts of Fock-exchange can in general be recommended for such ionic systems.
Co-reporter:Predrag V. Petrović, Stefan Grimme, Snežana D. Zarić, Michel Pfeffer and Jean-Pierre Djukic
Physical Chemistry Chemical Physics 2014 - vol. 16(Issue 28) pp:NaN14698-14698
Publication Date(Web):2014/06/02
DOI:10.1039/C4CP01500B
Self-aggregation in water of anti-cancer agents such as oxaliplatin (1) or its palladium-containing parent (2) is suspected to be the main reason for the exceptional resistance of concentrated infusions of these complexes to hydrolysis; this hypothesis, i.e. the self-association of metal chelates, was investigated in a systematic manner by experimental and theoretical means. 1H diffusion-ordered NMR spectroscopy (DOSY NMR) and UV-visible absorption titration were inconclusive as to the formation of a dimer of 1 in water or DMSO. Further isothermal titration calorimetry (ITC) methods allowed the accurate determination of the enthalpy of formation of only the homodimer [2]2 and putative heterodimer [1·2] together with an estimation of the formation constants, which indicate that dimer formation is not a spontaneous process in solution, whereas electrospray ESI mass spectroscopy tends to suggest the contrary in the gas phase. A dispersion-corrected DFT method, i.e. DFT-D (BLYP-D3), was used to model the aggregation in solution (COSMO) and to investigate the assisting role of London force in the cohesion of bimolecular aggregates. The concordance of experimental and theoretical thermodynamic parameters was judged reasonably even though the treatment of solvation by conventional continuum models does not account for specific interactions of the solute with molecules of solvent; nonetheless these results outline the importance of dispersion, a.k.a. London force. The role of the latter was further stressed by computing the affinities of 1 and 2 for the lipophilic cavity of cucurbit[7]uril in modeled water (COSMO-RS), which were preliminarily determined experimentally by ITC methods using pure water as solvent. From our investigations carried out in pure water the connection between the notorious chemical stability of “concentrated” infusions of 1 in aqueous media and the formation of oligomers remains unsettled.
Co-reporter:Tobias Risthaus, Andreas Hansen and Stefan Grimme
Physical Chemistry Chemical Physics 2014 - vol. 16(Issue 28) pp:
Publication Date(Web):
DOI:10.1039/C3CP54517B
Co-reporter:Stefan Grimme and Marc Steinmetz
Physical Chemistry Chemical Physics 2016 - vol. 18(Issue 31) pp:NaN20937-20937
Publication Date(Web):2015/12/03
DOI:10.1039/C5CP06600J
We present a revised form of a double hybrid density functional (DHDF) dubbed PWRB95. It contains semi-local Perdew–Wang exchange and Becke95 correlation with a fixed amount of 50% non-local Fock exchange. New features are that the robust random phase approximation (RPA) is used to calculate the non-local correlation part instead of a second-order perturbative treatment as in standard DHDF, and the non-self-consistent evaluation of the Fock exchange with KS-orbitals at the GGA level which leads to a significant reduction of the computational effort. To account for London dispersion effects we include the non-local VV10 dispersion functional. Only three empirical scaling parameters were adjusted. The PWRB95 results for extensive standard thermochemical benchmarks (GMTKN30 data base) are compared to those of well-known functionals from the classes of (meta-)GGAs, (meta-)hybrid functionals, and DHDFs, as well as to standard (direct) RPA. The new method is furthermore tested on prototype bond activations with (Ni/Pd)-based transition metal catalysts, and two difficult cases for DHDF, namely the isomerization reaction of the [Cu2(en)2O2]2+ complex and the singlet–triplet energy difference in highly unsaturated cyclacenes. The results show that PWRB95 is almost as accurate as standard DHDF for main-group thermochemistry but has a similar or better performance for non-covalent interactions, more difficult transition metal containing molecules and other electronically problematic cases. Because of its relatively weak basis set dependence, PWRB95 can be applied even in combination with AO basis sets of only triple-zeta quality which yields huge overall computational savings by a factor of about 40 compared to standard DHDF/‘quadruple-zeta’ calculations. Structure optimizations of small molecules with PWRB95 indicate an accurate description of bond distances superior to that provided by TPSS-D3, PBE0-D3, or other RPA type methods.
Co-reporter:Jan Gerit Brandenburg, Eike Caldeweyher and Stefan Grimme
Physical Chemistry Chemical Physics 2016 - vol. 18(Issue 23) pp:NaN15523-15523
Publication Date(Web):2016/05/24
DOI:10.1039/C6CP01697A
We extend the recently introduced PBEh-3c global hybrid density functional [S. Grimme et al., J. Chem. Phys., 2015, 143, 054107] by a screened Fock exchange variant based on the Henderson–Janesko–Scuseria exchange hole model. While the excellent performance of the global hybrid is maintained for small covalently bound molecules, its performance for computed condensed phase mass densities is further improved. Most importantly, a speed up of 30 to 50% can be achieved and especially for small orbital energy gap cases, the method is numerically much more robust. The latter point is important for many applications, e.g., for metal–organic frameworks, organic semiconductors, or protein structures. This enables an accurate density functional based electronic structure calculation of a full DNA helix structure on a single core desktop computer which is presented as an example in addition to comprehensive benchmark results.
Co-reporter:Christian Mück-Lichtenfeld and Stefan Grimme
Dalton Transactions 2012 - vol. 41(Issue 30) pp:NaN9118-9118
Publication Date(Web):2012/05/03
DOI:10.1039/C2DT30562C
The energy profiles of the activation reaction of small molecules (H2, Br2 and CO2) with boron/phosphorus frustrated Lewis pairs (FLPs) have been calculated with dispersion corrected DFT (TPSS-D3). We have investigated the cooperative nature of the reactions by analyzing interaction energies in the ternary system and for reactant pairs. The non-additive contributions to the total interaction energy add to the driving force of the activation reaction, even at early stages of the process. We propose the isosurface representation of the many-body deformation density Δρmb as a qualitative tool to visualize cooperative, non-additive effects in complex chemical systems.
Co-reporter:Ramesh C. Samanta, Suman De Sarkar, Roland Fröhlich, Stefan Grimme and Armido Studer
Chemical Science (2010-Present) 2013 - vol. 4(Issue 5) pp:NaN2184-2184
Publication Date(Web):2013/03/05
DOI:10.1039/C3SC00099K
This edge article reports the synthesis and full characterization including X-ray analysis of three different acylazolium ions. The reactivity of these acylazolium ions as acylating reagents of amines and alcohols is discussed. Whereas benzylamine slowly reacts with the acylazolium ions, benzyl alcohol acylation does not occur. However, upon activation of the alcohol with an N-heterocyclic carbene (NHC) as catalyst, efficient esterification is achieved. Importantly, benzylester formation is obtained in the presence of benzylamine upon selective alcohol activation by the NHC. High level DFT calculations reveal that alcohol activation occurs by strong H-bond formation between the NHC and the alcohol thereby increasing the nucleophilicity of the alcohol. For oxidatively generated acylazolium ions under NHC catalysis, the carbene has a dual role (cooperative catalysis): (a) the NHC is used for generation of the acylazolium ion and (b) the NHC is used for activation of the alcohol in the subsequent acylation step. NHC-catalyzed selective acylation of benzyl alcohol in the presence of benzylamine can also be achieved with trifluoroethyl and hexafluoroisopropylesters as acylation reagents. Moreover, an enol acetate also shows high O-selectivity as a chemoselective acetylation reagent. Kinetic and mechanistic studies are provided and some examples of the chemoselective acylation of amino alcohols are presented.
Co-reporter:Jens Antony, Rebecca Sure and Stefan Grimme
Chemical Communications 2015 - vol. 51(Issue 10) pp:NaN1774-1774
Publication Date(Web):2014/11/14
DOI:10.1039/C4CC06722C
A recently published theoretical approach employing a nondynamic structure model using dispersion-corrected density functional theory (DFT-D3) to calculate equilibrium free energies of association (Chem. Eur. J., 2012, 18, 9955–9964) is illustrated by its application to eight new supramolecular complexes. We compare with experimentally known binding constants which span the range from −3.3 to −20.3 kcal mol−1. The mean deviation of calculated from measured ΔGa results in 0.4 kcal mol−1, the mean absolute deviation in 1.8 kcal mol−1 excluding two outliers for which the computed solvation free energies are identified as the largest error source. A survey of previous applications of the theoretical approach and related computational studies is given underlining its good accuracy. It is concluded that structures and gas phase interaction energies can be computed routinely with good to high accuracy (relative errors for interaction energies of 5–10%) for complexes with about 200–300 atoms.
Co-reporter:Rebecca Sure and Stefan Grimme
Chemical Communications 2016 - vol. 52(Issue 64) pp:NaN9896-9896
Publication Date(Web):2016/07/07
DOI:10.1039/C6CC03664C
Recently, Diederich et al. synthesized the first supramolecular capsule with a well-defined four-point halogen bonding interaction [Angew. Chem., Int. Ed., 2015, 54, 12339]. This interesting system comprising about 400 atoms represents a challenging test case for accurate quantum chemical methods. We investigate it with our new density functional based composite method for structures and noncovalent interactions (PBEh-3c) as well as our standard protocol for supramolecular thermochemistry and give predictions for chemical modifications to improve the binding strength.
Co-reporter:Christophe Werlé, Sebastian Dohm, Corinne Bailly, Lydia Karmazin, Louis Ricard, Nicolas Sieffert, Michel Pfeffer, Andreas Hansen, Stefan Grimme and Jean-Pierre Djukic
Dalton Transactions 2017 - vol. 46(Issue 25) pp:NaN8137-8137
Publication Date(Web):2017/05/17
DOI:10.1039/C7DT00872D
Kinetically unstable heteroleptic trans-bispalladacycles were isolated by using hemichelation. Three structures of trans isomers and five of cis isomers were characterized by X-ray diffraction analysis. The ready trans-to-cis isomerization of such hemichelates that was monitored by variable temperature NMR experiments is facilitated dynamically because the Pd(II) center can preserve its square planar coordination in a rather low lying transition state, which was localized by methods of the density functional theory. This process is not achievable in the isomerization of conventional homoleptic trans-bispalladacycles since it involves the preliminary partial chelate decoordination and an unfavorable high-lying planar trigonal coordinated – or Y-shaped-Pd(II) transition state according to DFT investigations.
Co-reporter:A. W. Kyri, R. Kunzmann, G. Schnakenburg, Z.-W. Qu, S. Grimme and R. Streubel
Chemical Communications 2016 - vol. 52(Issue 91) pp:NaN13364-13364
Publication Date(Web):2016/10/26
DOI:10.1039/C6CC07081G
Despite intense research in FLP chemistry, nothing is known about monomolecular anionic FLPs and/or complexes thereof. Herein, synthesis and reaction of the first anionic FLP complex is described using [(OC)5W{(Me3Si)2HCP(H)OLi(12-crown-4)}], Cy2BCl and subsequent deprotonation by KHMDS. The obtained anionic FLP complex reacts readily with CO2 in a concerted manner.
Co-reporter:Vilhjálmur Ásgeirsson, Christoph A. Bauer and Stefan Grimme
Physical Chemistry Chemical Physics 2016 - vol. 18(Issue 45) pp:NaN31026-31026
Publication Date(Web):2016/10/19
DOI:10.1039/C6CP06180J
We present negative ion-mode simulations within the QCEIMS program [Grimme, Angew. Chem., Int. Ed., 2013, 52, 6306]. It is an exhaustive and robust ab initio molecular dynamics/stochastic algorithm used to perform simulations of unimolecular decomposition of anions, in unprecedented detail. The objective of this approach is to compliment electron attachment spectroscopy and aid in the interpretation of relevant dissociation dynamics. Prototypical simulations are performed for the four nitrile compounds acetonitrile, cyanamide, aminoacetonitrile, and trifluoroacetonitrile. The unique decomposition pathways which naturally occur in the simulations are addressed along with fractional yields, reaction times and relative intensities of the fragments. Furthermore, trajectories of selected decomposition pathways of the aminoacetonitrile anion are investigated in greater detail, where we find that the relevant HOMO of the anion has a mixed π* and σ* character delocalized over the entire molecule.
Co-reporter:Stefan Grimme and Marc Steinmetz
Physical Chemistry Chemical Physics 2013 - vol. 15(Issue 38) pp:NaN16042-16042
Publication Date(Web):2013/07/11
DOI:10.1039/C3CP52293H
A benchmark set of 25 rotational constants measured in the gas phase for nine molecules (termed ROT25) was compiled from available experimental data. The medium-sized molecules with 18–35 atoms cover common (bio)organic structure motifs including hydrogen bonding and flexible side chains. They were each considered in a single conformation. The experimental B0 values were back-corrected to reference equilibrium rotational constants (Be) by computation of the vibrational corrections ΔBvib. Various density functional theory (DFT) methods and Hartree–Fock with and without dispersion corrections as well as MP2 type methods and semi-empirical quantum chemical approaches are investigated. The ROT25 benchmark tests their ability to describe covalent bond lengths, longer inter-atomic distances, and the relative orientation of functional groups (intramolecular non-covalent interactions). In general, dispersion corrections to DFT and HF increase Be values (shrink molecular size) significantly by about 0.5–1.5% thereby in general improving agreement with the reference data. Regarding DFT methods, the overall accuracy of the optimized structures roughly follows the ‘Jacobs ladder’ classification scheme, i.e., it decreases in the series double-hybrid > (meta)hybrid > (meta)GGA > LDA. With B2PLYP-D3, SCS-MP2, B3LYP-D3/NL, or PW6B95-D3 methods and extended QZVP (def2-TZVP) AO basis sets, Be values, accurate to about 0.3–0.6 (0.5–1)% on average, can be computed routinely. The accuracy of B2PLYP-D3/QZVP with a mean deviation of only 3 MHz and a standard deviation of 0.24% is exceptional and we recommend this method when highly accurate structures are required or for problematic conformer assignments. The correlation effects for three inter-atomic distance regimes (covalent, medium-range, long) and the performance of minimal basis set (semi-empirical) methods are discussed.

![Spiro[5.6]dodecan-8-ol](http://img.cochemist.com/ccimg/928900/928822-25-7.png)
![Spiro[5.6]dodecan-8-ol](http://img.cochemist.com/ccimg/928900/928822-25-7_b.png)

![1H-Cyclopenta[c]furan, hexahydro-1,1-dimethyl-, (3aR,6aS)-rel-](http://img.cochemist.com/ccimg/928900/928822-21-3.png)
![1H-Cyclopenta[c]furan, hexahydro-1,1-dimethyl-, (3aR,6aS)-rel-](http://img.cochemist.com/ccimg/928900/928822-21-3_b.png)


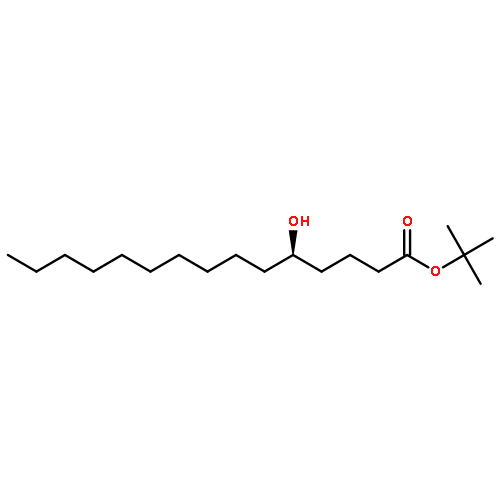
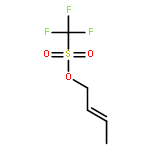
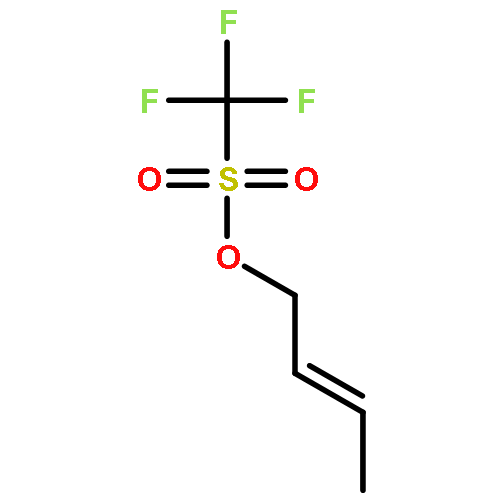
![1-Oxaspiro[2.5]octane, 4-[(2Z)-2-hexenyloxy]-](http://img.cochemist.com/ccimg/737800/737772-34-8.png)
![1-Oxaspiro[2.5]octane, 4-[(2Z)-2-hexenyloxy]-](http://img.cochemist.com/ccimg/737800/737772-34-8_b.png)
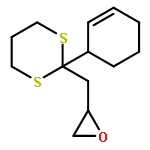
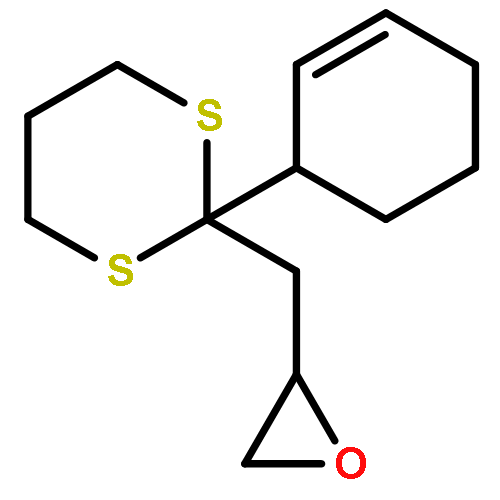

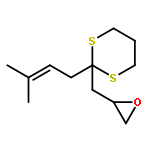
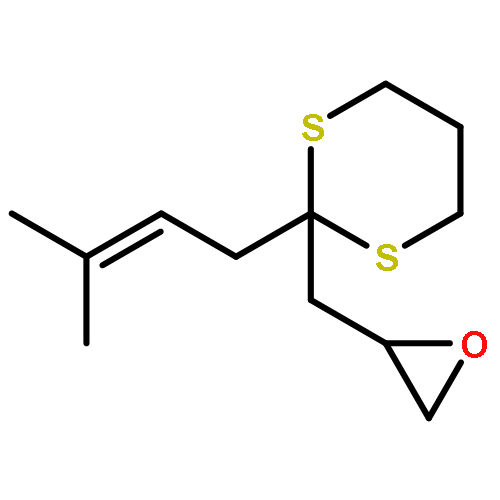


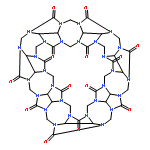
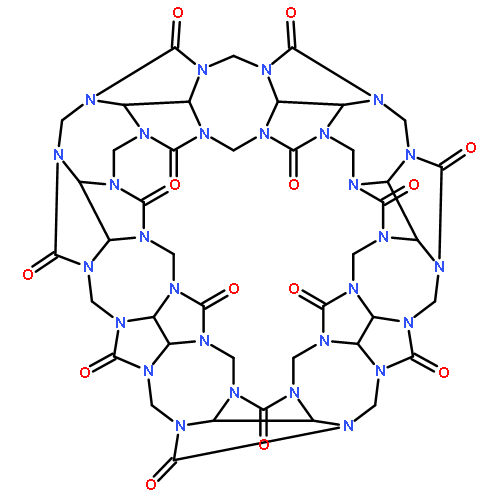
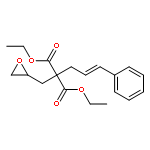
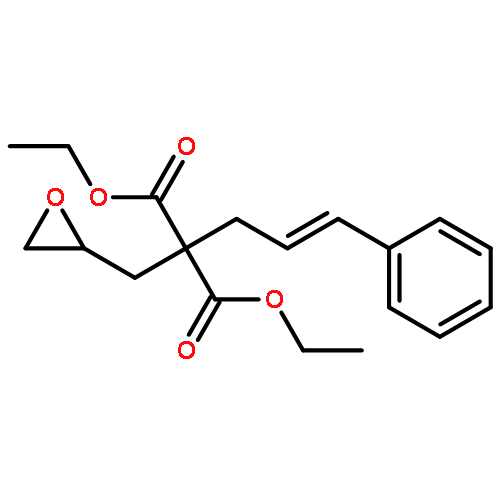

![3-Tosyl-6-oxa-3-azabicyclo[3.1.0]hexane](http://img.cochemist.com/ccimg/159600/159555-66-5.png)
![3-Tosyl-6-oxa-3-azabicyclo[3.1.0]hexane](http://img.cochemist.com/ccimg/159600/159555-66-5_b.png)
![Tricyclo[3.3.1.13,7]decane-1,3-dimethanethiol](http://img.cochemist.com/ccimg/151200/151133-08-3.png)
![Tricyclo[3.3.1.13,7]decane-1,3-dimethanethiol](http://img.cochemist.com/ccimg/151200/151133-08-3_b.png)
![Benzo[o]bistriphenyleno[2,1,12,11-efghi:2',1',12',11'-uvabc]ovalene](http://img.cochemist.com/ccimg/123200/123178-24-5.png)
![Benzo[o]bistriphenyleno[2,1,12,11-efghi:2',1',12',11'-uvabc]ovalene](http://img.cochemist.com/ccimg/123200/123178-24-5_b.png)
![1-Piperidinyloxy, 4-[[(cyclohexylamino)thioxomethyl]amino]-2,2,6,6-tetramethyl-](http://img.cochemist.com/ccimg/111200/111103-86-7.png)
![1-Piperidinyloxy, 4-[[(cyclohexylamino)thioxomethyl]amino]-2,2,6,6-tetramethyl-](http://img.cochemist.com/ccimg/111200/111103-86-7_b.png)
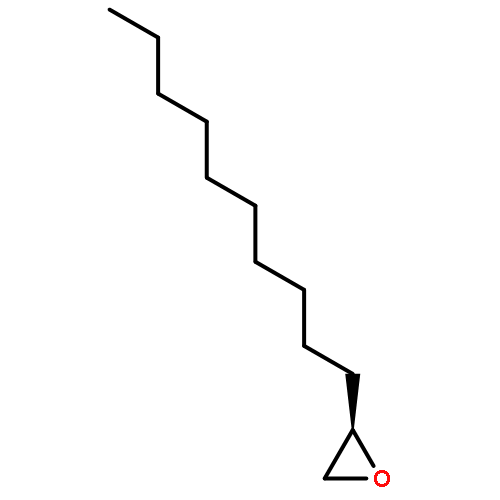
![Spiro[5.6]dodec-7-ene](http://img.cochemist.com/ccimg/105900/105838-48-0.png)
![Spiro[5.6]dodec-7-ene](http://img.cochemist.com/ccimg/105900/105838-48-0_b.png)




![1-Oxaspiro[3.5]nonane, 7-(1,1-dimethylethyl)-](http://img.cochemist.com/ccimg/87600/87597-09-9.png)
![1-Oxaspiro[3.5]nonane, 7-(1,1-dimethylethyl)-](http://img.cochemist.com/ccimg/87600/87597-09-9_b.png)




![1H,5H,11H-[1]Benzopyrano[6,7,8-ij]quinolizin-11-one, 2,3,6,7-tetrahydro-9-(trifluoromethyl)-](/data/chemimg/905400/53518-18-6.png)
![1H,5H,11H-[1]Benzopyrano[6,7,8-ij]quinolizin-11-one, 2,3,6,7-tetrahydro-9-(trifluoromethyl)-](/data/chemimg/905400/53518-18-6_b.png)
![1-Piperidinyloxy,4-[(cyclohexylcarbonimidoyl)amino]-2,2,6,6-tetramethyl-](http://img.cochemist.com/ccimg/42300/42249-40-1.png)
![1-Piperidinyloxy,4-[(cyclohexylcarbonimidoyl)amino]-2,2,6,6-tetramethyl-](http://img.cochemist.com/ccimg/42300/42249-40-1_b.png)

![2-[(4-METHYL-2-NITROPHENYL)DIAZENYL]-N-(2-METHYLPHENYL)-3-OXOBUTA<WBR />NAMIDE](http://img.cochemist.com/ccimg/36300/36291-48-2.png)
![2-[(4-METHYL-2-NITROPHENYL)DIAZENYL]-N-(2-METHYLPHENYL)-3-OXOBUTA<WBR />NAMIDE](http://img.cochemist.com/ccimg/36300/36291-48-2_b.png)
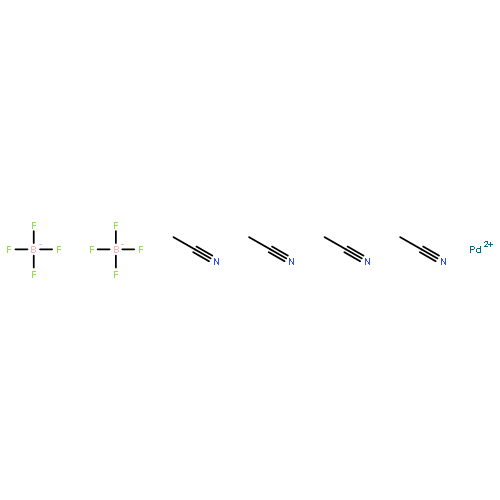
![[6-(HYDROXYMETHYL)PYRIDIN-3-YL]METHANOL](/data/chemimg/417700/21514-99-8.png)
![[6-(HYDROXYMETHYL)PYRIDIN-3-YL]METHANOL](/data/chemimg/417700/21514-99-8_b.png)
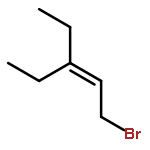
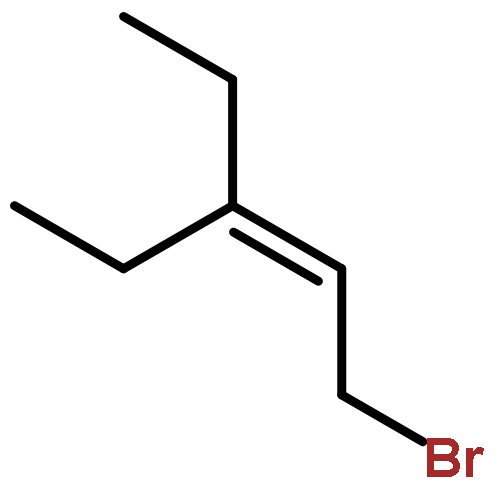

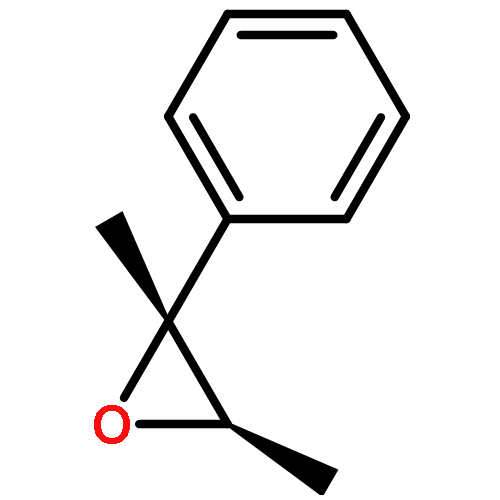
![2,2,2-Trifluoro-N-[3-(trifluoroacetamido)phenyl]-acetamide](http://img.cochemist.com/ccimg/15100/15059-42-4.png)
![2,2,2-Trifluoro-N-[3-(trifluoroacetamido)phenyl]-acetamide](http://img.cochemist.com/ccimg/15100/15059-42-4_b.png)
![5,16:8,13-Diethenodibenzo[a,g]cyclododecene,6,7,14,15-tetrahydro-, stereoisomer (9CI)](http://img.cochemist.com/ccimg/14800/14724-91-5.png)
![5,16:8,13-Diethenodibenzo[a,g]cyclododecene,6,7,14,15-tetrahydro-, stereoisomer (9CI)](http://img.cochemist.com/ccimg/14800/14724-91-5_b.png)




![Carbazolo[3,4-c]carbazole, 5,10-dihydro-](http://img.cochemist.com/ccimg/7300/7259-13-4.png)
![Carbazolo[3,4-c]carbazole, 5,10-dihydro-](http://img.cochemist.com/ccimg/7300/7259-13-4_b.png)
![Bicyclo[2.2.0]hexa-2,5-diene](http://img.cochemist.com/ccimg/5700/5649-95-6.png)
![Bicyclo[2.2.0]hexa-2,5-diene](http://img.cochemist.com/ccimg/5700/5649-95-6_b.png)
![N'-(3-AMINOPROPYL)-N'-[2-[BIS(3-AMINOPROPYL)AMINO]ETHYL]PROPANE-1,3-DIAMINE](http://img.cochemist.com/ccimg/4900/4879-98-5.png)
![N'-(3-AMINOPROPYL)-N'-[2-[BIS(3-AMINOPROPYL)AMINO]ETHYL]PROPANE-1,3-DIAMINE](http://img.cochemist.com/ccimg/4900/4879-98-5_b.png)
![6-phenyl-7-oxabicyclo[4.1.0]heptane](http://img.cochemist.com/ccimg/4900/4829-01-0.png)
![6-phenyl-7-oxabicyclo[4.1.0]heptane](http://img.cochemist.com/ccimg/4900/4829-01-0_b.png)
![Bicyclo[2.2.1]hept-2-ene, 2-phenyl-](http://img.cochemist.com/ccimg/4300/4237-08-5.png)
![Bicyclo[2.2.1]hept-2-ene, 2-phenyl-](http://img.cochemist.com/ccimg/4300/4237-08-5_b.png)
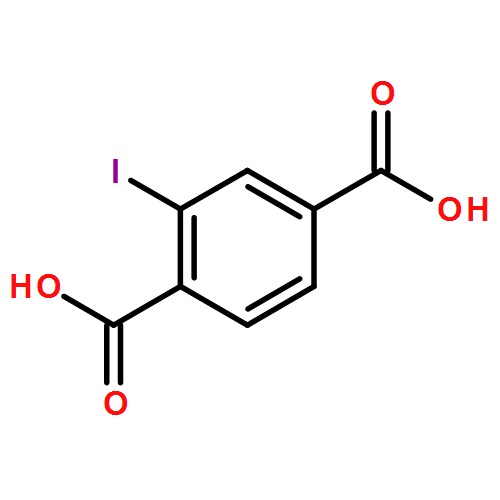
![5,12[1',2']:6,11[1'',2'']-Dibenzenodibenzo[a,e]cyclooctene, 5,6,11,12-tetrahydro-](/data/chemimg/924600/1627-06-1.png)
![5,12[1',2']:6,11[1'',2'']-Dibenzenodibenzo[a,e]cyclooctene, 5,6,11,12-tetrahydro-](/data/chemimg/924600/1627-06-1_b.png)
![Bicyclo[2.2.1]heptan-2-one,1,3,3-trimethyl-](http://img.cochemist.com/ccimg/1200/1195-79-5.png)
![Bicyclo[2.2.1]heptan-2-one,1,3,3-trimethyl-](http://img.cochemist.com/ccimg/1200/1195-79-5_b.png)
![Spiro[5.6]dodecan-7-ol](http://img.cochemist.com/ccimg/1200/1130-20-7.png)
![Spiro[5.6]dodecan-7-ol](http://img.cochemist.com/ccimg/1200/1130-20-7_b.png)






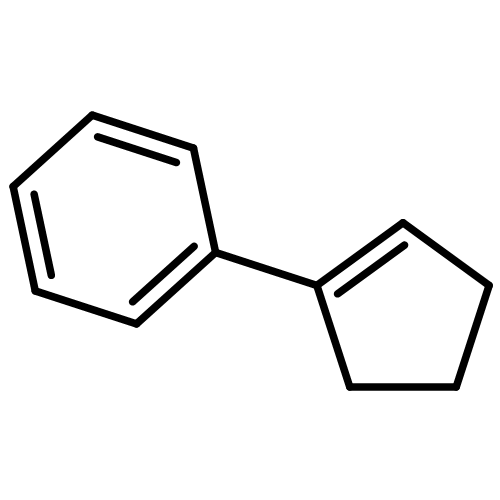


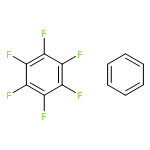
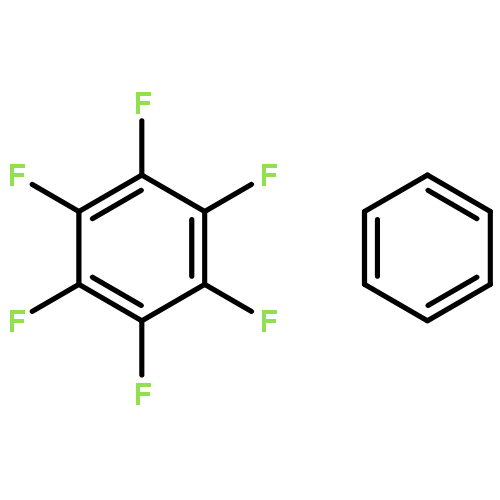
![Tetracyclo[2.2.0.02,6.03,5]hexane](http://img.cochemist.com/ccimg/700/650-42-0.png)
![Tetracyclo[2.2.0.02,6.03,5]hexane](http://img.cochemist.com/ccimg/700/650-42-0_b.png)
![tribenzo[fg,ij,rst]pentaphene](http://img.cochemist.com/ccimg/200/190-81-8.png)
![tribenzo[fg,ij,rst]pentaphene](http://img.cochemist.com/ccimg/200/190-81-8_b.png)
![Hexabenzo[bc,ef,hi,kl,no,qr]coronene](/data/chemimg/1472400/190-24-9.png)
![Hexabenzo[bc,ef,hi,kl,no,qr]coronene](/data/chemimg/1472400/190-24-9_b.png)
![Phenanthro[3,4-c]phenanthrene](http://img.cochemist.com/ccimg/200/187-83-7.png)
![Phenanthro[3,4-c]phenanthrene](http://img.cochemist.com/ccimg/200/187-83-7_b.png)




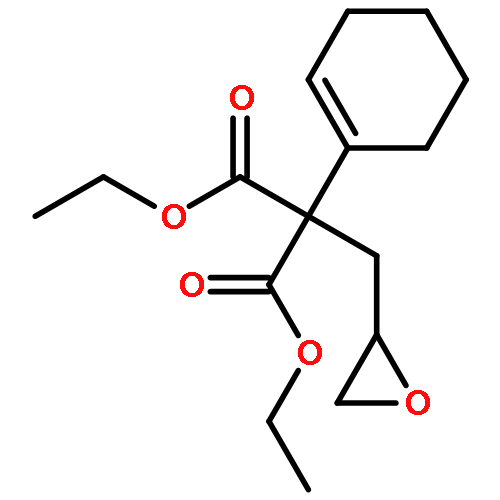
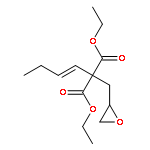
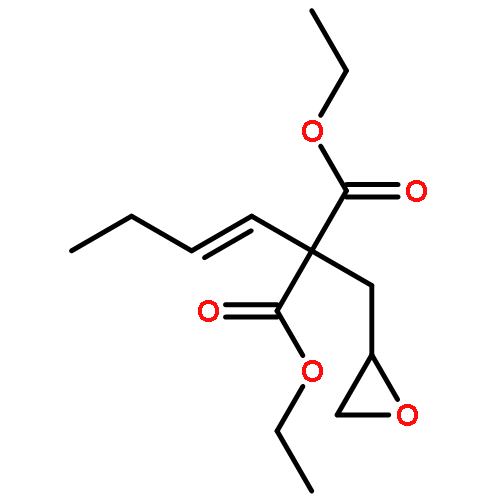
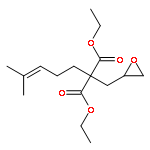
![3-iodo-1-[3-iodo-2-(methoxymethoxy)naphthalen-1-yl]-2-(methoxymethoxy)naphthalene](http://img.cochemist.com/ccimg/219600/219583-87-6.png)
![3-iodo-1-[3-iodo-2-(methoxymethoxy)naphthalen-1-yl]-2-(methoxymethoxy)naphthalene](http://img.cochemist.com/ccimg/219600/219583-87-6_b.png)
![Titanium,dichlorobis[(1,2,3,4,5-h)-1-methyl-2,4-cyclopentadien-1-yl]- (9CI)](http://img.cochemist.com/ccimg/1300/1282-40-2.png)
![Titanium,dichlorobis[(1,2,3,4,5-h)-1-methyl-2,4-cyclopentadien-1-yl]- (9CI)](http://img.cochemist.com/ccimg/1300/1282-40-2_b.png)