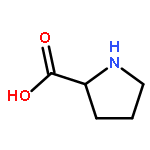
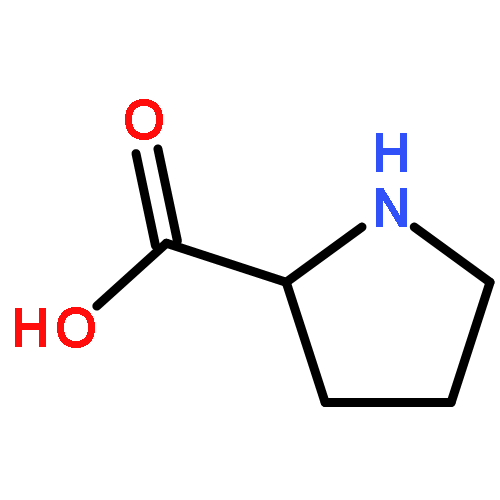
Co-reporter:Anil P. Jagtap, Michel-Andreas Geiger, Daniel Stöppler, Marcella Orwick-Rydmark, Hartmut Oschkinat and Snorri Th. Sigurdsson
Chemical Communications 2016 vol. 52(Issue 43) pp:7020-7023
Publication Date(Web):10 May 2016
DOI:10.1039/C6CC01813K
Dynamic nuclear polarization (DNP) is an efficient method to overcome the inherent low sensitivity of magic-angle spinning (MAS) solid-state NMR. We report a new polarizing agent (bcTol), designed for biological applications, that yielded an enhancement value of 244 in a microcrystalline SH3 domain sample at 110 K.
Co-reporter:Sascha Lange;W. Trent Franks;Nandhakishore Rajagopalan;Kristina Döring;Michel A. Geiger;Arne Linden;Barth-Jan van Rossum;Günter Kramer;Bernd Bukau
Science Advances 2016 Vol 2(8) pp:e1600379
Publication Date(Web):19 Aug 2016
DOI:10.1126/sciadv.1600379
DNP-enhanced MAS NMR reveals extended conformations for the DsbA signal peptide within the ribosome exit tunnel.
Co-reporter:Sara Bruun, Daniel Stoeppler, Anke Keidel, Uwe Kuhlmann, Meike Luck, Anne Diehl, Michel-Andreas Geiger, David Woodmansee, Dirk Trauner, Peter Hegemann, Hartmut Oschkinat, Peter Hildebrandt, and Katja Stehfest
Biochemistry 2015 Volume 54(Issue 35) pp:
Publication Date(Web):August 3, 2015
DOI:10.1021/acs.biochem.5b00597
Channelrhodopsins (ChR) are light-gated ion channels of green algae that are widely used to probe the function of neuronal cells with light. Most ChRs show a substantial reduction in photocurrents during illumination, a process named “light adaptation”. The main objective of this spectroscopic study was to elucidate the molecular processes associated with light–dark adaptation. Here we show by liquid and solid-state nuclear magnetic resonance spectroscopy that the retinal chromophore of fully dark-adapted ChR is exclusively in an all-trans configuration. Resonance Raman (RR) spectroscopy, however, revealed that already low light intensities establish a photostationary equilibrium between all-trans,15-anti and 13-cis,15-syn configurations at a ratio of 3:1. The underlying photoreactions involve simultaneous isomerization of the C(13)═C(14) and C(15)═N bonds. Both isomers of this DAapp state may run through photoinduced reaction cycles initiated by photoisomerization of only the C(13)═C(14) bond. RR spectroscopic experiments further demonstrated that photoinduced conversion of the apparent dark-adapted (DAapp) state to the photocycle intermediates P500 and P390 is distinctly more efficient for the all-trans isomer than for the 13-cis isomer, possibly because of different chromophore–water interactions. Our data demonstrating two complementary photocycles of the DAapp isomers are fully consistent with the existence of two conducting states that vary in quantitative relation during light–dark adaptation, as suggested previously by electrical measurements.
Co-reporter:Dr. Carolyn Vargas;Dr. Gerald Radziwill;Dr. Gerd Krause;Dr. Anne Diehl;Dr. Sro Keller;Dr. Nestor Kamdem;Dr. Constantin Czekelius;Annika Kreuchwig;Dr. Peter Schmieder;Dr. Declan Doyle;Dr. Karin Moelling;Dr. Volker Hagen;Dr. Markus Schade;Dr. Hartmut Oschkinat
ChemMedChem 2014 Volume 9( Issue 7) pp:1458-1462
Publication Date(Web):
DOI:10.1002/cmdc.201300553
Abstract
PDZ (PSD-95, Dlg, ZO-1) domains are ubiquitous interaction modules that are involved in many cellular signal transduction pathways. Interference with PDZ-mediated protein–protein interactions has important implications in disease-related signaling processes. For this reason, PDZ domains have gained attention as potential targets for inhibitor design and, in the long run, drug development. Herein we report the development of small molecules to probe the function of the PDZ domain from human AF6 (ALL1-fused gene from chromosome 6), which is an essential component of cell–cell junctions. These compounds bind to AF6 PDZ with substantially higher affinity than the peptide (Ile-Gln-Ser-Val-Glu-Val) derived from its natural ligand, EphB2. In intact cells, the compounds inhibit the AF6–Bcr interaction and interfere with epidermal growth factor (EGF)-dependent signaling.
Co-reporter:Dr. Ümit Akbey;Dr. Andrew J. Nieuwkoop;Dr. Sebastian Wegner;Anja Voreck;Dr. Britta Kunert;Dr. Priyanga Bara;Dr. Frank Engelke;Dr. Niels Chr. Nielsen;Dr. Hartmut Oschkinat
Angewandte Chemie International Edition 2014 Volume 53( Issue 9) pp:2438-2442
Publication Date(Web):
DOI:10.1002/anie.201308927
Abstract
1H-detected magic-angle spinning NMR experiments facilitate structural biology of solid proteins, which requires using deuterated proteins. However, often amide protons cannot be back-exchanged sufficiently, because of a possible lack of solvent exposure. For such systems, using 2H excitation instead of 1H excitation can be beneficial because of the larger abundance and shorter longitudinal relaxation time, T1, of deuterium. A new structure determination approach, “quadruple-resonance NMR spectroscopy”, is presented which relies on an efficient 2H-excitation and 2H-13C cross-polarization (CP) step, combined with 1H detection. We show that by using 2H-excited experiments better sensitivity is possible on an SH3 sample recrystallized from 30 % H2O. For a membrane protein, the ABC transporter ArtMP in native lipid bilayers, different sets of signals can be observed from different initial polarization pathways, which can be evaluated further to extract structural properties.
Co-reporter:Sascha Lange, Arne H. Linden, Ümit Akbey, W. Trent Franks, Nikolaus M. Loening, Barth-Jan van Rossum, Hartmut Oschkinat
Journal of Magnetic Resonance 2012 216() pp: 209-212
Publication Date(Web):
DOI:10.1016/j.jmr.2012.01.002
Co-reporter:Ümit Akbey, Barth-Jan van Rossum, Hartmut Oschkinat
Journal of Magnetic Resonance 2012 217() pp: 77-85
Publication Date(Web):
DOI:10.1016/j.jmr.2012.02.015
Co-reporter:Dr. Benjamin Bardiaux;Dr. Barth-Jan vanRossum; Michael Nilges; Hartmut Oschkinat
Angewandte Chemie International Edition 2012 Volume 51( Issue 28) pp:6916-6919
Publication Date(Web):
DOI:10.1002/anie.201201783
Co-reporter:Arne H. Linden ; Sascha Lange ; W. Trent Franks ; Ümit Akbey ; Edgar Specker ; Barth-Jan van Rossum
Journal of the American Chemical Society 2011 Volume 133(Issue 48) pp:19266-19269
Publication Date(Web):October 31, 2011
DOI:10.1021/ja206999c
Methods enabling structural studies of membrane-integrated receptor systems without the necessity of purification provide an attractive perspective in membrane protein structural and molecular biology. This has become feasible in principle since the advent of dynamic nuclear polarization (DNP) magic-angle-spinning NMR spectroscopy, which delivers the required sensitivity. In this pilot study, we observed well-resolved solid-state NMR spectra of extensively 13C-labeled neurotoxin II bound to the nicotinic acetylcholine receptor (nAChR) in native membranes. We show that TOTAPOL, a biradical required for DNP, is localized at membrane and protein surfaces. The concentration of active, membrane-attached biradical decreases with time, probably because of reactive components of the membrane preparation. An optimal distribution of active biradical has strong effects on the NMR data. The presence of inactive TOTAPOL in membrane-proximal situations but active biradical in the surrounding water/glycerol “glass” leads to well-resolved spectra, yet a considerable enhancement (ε = 12) is observed. The resulting spectra of a protein ligand bound to its receptor are paving the way for further DNP investigations of proteins embedded in native membrane patches.
Co-reporter:Dr. Ümit Akbey;Francesca Camponeschi;Dr. Barth-Jan van Rossum ; Hartmut Oschkinat
ChemPhysChem 2011 Volume 12( Issue 11) pp:2092-2096
Publication Date(Web):
DOI:10.1002/cphc.201100084
Co-reporter:Dr. Rasmus Linser;Muralidhar Dasari;Dr. Matthias Hiller;Dr. Victoria Higman;Uwe Fink;Dr. Juan-Miguel LopezdelAmo;Stefan Markovic;Liselotte Hel;Brigitte Kessler;Dr. Peter Schmieder;Dr. Dieter Oesterhelt;Dr. Hartmut Oschkinat;Dr. Bernd Reif
Angewandte Chemie 2011 Volume 123( Issue 19) pp:4601-4605
Publication Date(Web):
DOI:10.1002/ange.201008244
Co-reporter:Dr. Rasmus Linser;Muralidhar Dasari;Dr. Matthias Hiller;Dr. Victoria Higman;Uwe Fink;Dr. Juan-Miguel LopezdelAmo;Stefan Markovic;Liselotte Hel;Brigitte Kessler;Dr. Peter Schmieder;Dr. Dieter Oesterhelt;Dr. Hartmut Oschkinat;Dr. Bernd Reif
Angewandte Chemie 2011 Volume 123( Issue 19) pp:
Publication Date(Web):
DOI:10.1002/ange.201101732
Co-reporter:Dr. Rasmus Linser;Muralidhar Dasari;Dr. Matthias Hiller;Dr. Victoria Higman;Uwe Fink;Dr. Juan-Miguel LopezdelAmo;Stefan Markovic;Liselotte Hel;Brigitte Kessler;Dr. Peter Schmieder;Dr. Dieter Oesterhelt;Dr. Hartmut Oschkinat;Dr. Bernd Reif
Angewandte Chemie International Edition 2011 Volume 50( Issue 19) pp:
Publication Date(Web):
DOI:10.1002/anie.201101732
Co-reporter:Dr. Rasmus Linser;Muralidhar Dasari;Dr. Matthias Hiller;Dr. Victoria Higman;Uwe Fink;Dr. Juan-Miguel LopezdelAmo;Stefan Markovic;Liselotte Hel;Brigitte Kessler;Dr. Peter Schmieder;Dr. Dieter Oesterhelt;Dr. Hartmut Oschkinat;Dr. Bernd Reif
Angewandte Chemie International Edition 2011 Volume 50( Issue 19) pp:4508-4512
Publication Date(Web):
DOI:10.1002/anie.201008244
Co-reporter:Stefan Jehle;Breanna S. Vollmar;Benjamin Bardiaux;Ponni Rajagopal;Katja K. Dove;Tamir Gonen;Rachel E. Klevit
PNAS 2011 Volume 108 (Issue 16 ) pp:6409-6414
Publication Date(Web):2011-04-19
DOI:10.1073/pnas.1014656108
The small heat shock protein (sHSP) αB-crystallin (αB) plays a key role in the cellular protection system against stress.
For decades, high-resolution structural studies on heterogeneous sHSPs have been confounded by the polydisperse nature of
αB oligomers. We present an atomic-level model of full-length αB as a symmetric 24-subunit multimer based on solid-state NMR,
small-angle X-ray scattering (SAXS), and EM data. The model builds on our recently reported structure of the homodimeric α-crystallin
domain (ACD) and C-terminal IXI motif in the context of the multimer. A hierarchy of interactions contributes to build multimers
of varying sizes: Interactions between two ACDs define a dimer, three dimers connected by their C-terminal regions define
a hexameric unit, and variable interactions involving the N-terminal region define higher-order multimers. Within a multimer,
N-terminal regions exist in multiple environments, contributing to the heterogeneity observed by NMR. Analysis of SAXS data
allows determination of a heterogeneity parameter for this type of system. A mechanism of multimerization into higher-order
asymmetric oligomers via the addition of up to six dimeric units to a 24-mer is proposed. The proposed asymmetric multimers
explain the homogeneous appearance of αB in negative-stain EM images and the known dynamic exchange of αB subunits. The model
of αB provides a structural basis for understanding known disease-associated missense mutations and makes predictions concerning
substrate binding and the reported fibrilogenesis of αB.
Co-reporter:Dr. Ümit Akbey;Dr. W. Trent Franks;Arne Linden;Sascha Lange; Robert G. Griffin;Dr. Barth-Jan vanRossum; Hartmut Oschkinat
Angewandte Chemie 2010 Volume 122( Issue 42) pp:7971-7974
Publication Date(Web):
DOI:10.1002/ange.201002044
Co-reporter:Vivien Lange Dr.;Johanna Becker-Baldus Dr.;Britta Kunert;Barth-Jan van Rossum Dr.;Fabio Casagre Dr.;Andreas Engel ;Yvette Roske Dr.;Frank M. Scheffel Dr.;Erwin Schneider
ChemBioChem 2010 Volume 11( Issue 4) pp:547-555
Publication Date(Web):
DOI:10.1002/cbic.200900472
Abstract
ATP-binding cassette (ABC) transport systems facilitate the translocation of substances, like amino acids, across cell membranes energised by ATP hydrolysis. This work describes first structural studies on the ABC transporter ArtMP from Geobacillus stearothermophilus in native lipid environment by magic-angle spinning NMR spectroscopy. The 2D crystals of ArtMP and 3D crystals of isolated ArtP were prepared in different nucleotide-bound or -unbound states. From selectively 13C,15N-labelled ArtP, several sequence-specific assignments were obtained, most of which could be transferred to spectra of ArtMP. Residues Tyr133 and Pro134 protrude directly into the ATP-binding pocket at the interface of the ArtP subunits, and hence, are sensitive monitors for structural changes during nucleotide binding and hydrolysis. Distinct sets of NMR shifts were obtained for ArtP with different phosphorylation states of the ligand. Indications were found for an asymmetric or inhomogeneous state of the ArtP dimer bound with triphosphorylated nucleotides. With this investigation, a model system was established for screening all functional states occurring in one ABC transporter in native lipid environment.
Co-reporter:Dr. Ümit Akbey;Dr. W. Trent Franks;Arne Linden;Sascha Lange; Robert G. Griffin;Dr. Barth-Jan vanRossum; Hartmut Oschkinat
Angewandte Chemie International Edition 2010 Volume 49( Issue 42) pp:7803-7806
Publication Date(Web):
DOI:10.1002/anie.201002044
Co-reporter:Johanna Becker;Neil Ferguson Dr.;Jeremy Flinders Dr.;Barth-Jan van Rossum Dr.;Alan R. Fersht
ChemBioChem 2008 Volume 9( Issue 12) pp:1946-1952
Publication Date(Web):
DOI:10.1002/cbic.200700706
Abstract
The second WW domain (WW2) of CA150, a human transcriptional activator, forms amyloid fibrils in vitro under physiological conditions. Based on experimental constraints from MAS NMR spectroscopy experiments, alanine scanning and electron microscopy, a structural model of CA150.WW2 amyloid fibrils was calculated earlier. Here, the assignment strategy is presented and suggested as a general approach for proteins that show intermediate line width. The 13C,13C correlation experiments were recorded on fully or partially 13C-labelled fibrils. The earlier 13C assignment (26 residues) was extended to 34 of the 40 residues by direct 13C-excitation experiments by using a deuterated sample that showed strongly improved line width. A 3D HNC-TEDOR (transferred-echo double-resonance) experiment with deuterated CA150.WW2 fibrils yielded 14 amide nitrogen and proton resonance assignments. The obtained chemical shifts were compared with the chemical shifts determined with the natively folded WW domain. TALOS (Torsion angle likelihood obtained from shift and sequence similarity) predictions confirmed that, under physiological conditions, the fibrillar form of CA150.WW2 adopts a significantly different β structure than the native WW-domain fold.
Co-reporter:Johanna Becker;Neil Ferguson;Henning Tidow;Sandra Tremmel;Timothy D. Sharpe;Gerd Krause;Jeremy Flinders;Miriana Petrovich;John Berriman;Alan R. Fersht
PNAS 2006 Volume 103 (Issue 44 ) pp:16248-16253
Publication Date(Web):2006-10-31
DOI:10.1073/pnas.0607815103
Human CA150, a transcriptional activator, binds to and is co-deposited with huntingtin during Huntington's disease. The second
WW domain of CA150 is a three-stranded β-sheet that folds in vitro in microseconds and forms amyloid fibers under physiological conditions. We found from exhaustive alanine scanning studies
that fibrillation of this WW domain begins from its denatured conformations, and we identified a subset of residues critical
for fibril formation. We used high-resolution magic-angle-spinning NMR studies on site-specific isotopically labeled fibrils
to identify abundant long-range interactions between side chains. The distribution of critical residues identified by the
alanine scanning and NMR spectroscopy, along with the electron microscopy data, revealed the protofilament repeat unit: a
26-residue nonnative β-hairpin. The structure we report has similarities to the hairpin formed by the Aβ
(1–40) protofilament, yet also contains closely packed side-chains in a “steric zipper” arrangement found in the cross-β spine formed
from small peptides from the Sup35 prion protein. Fibrillation of unrelated amyloidogenic sequences shows the common feature
of zippered repeat units that act as templates for fiber elongation.
Co-reporter:Mangesh Joshi;Carolyn Vargas;Prisca Boisguerin Dr.;Annette Diehl Dr.;Gerd Krause Dr.;Peter Schmieder Dr.;Karin Moelling Dr.;Volker Hagen Dr.;Markus Schade Dr. Dr.
Angewandte Chemie International Edition 2006 Volume 45(Issue 23) pp:
Publication Date(Web):3 MAY 2006
DOI:10.1002/anie.200503965
In the groove: The “drugability” of protein–protein interaction domains is still a matter of debate. The 3D structure of a complex of a small organic ligand and the AF6 PDZ domain revealed the creation of a binding pocket by the ligand (see picture). The derived compound is able to compete with a natural peptide ligand of the domain and represents a basic building block for the generation of selective PDZ inhibitors.
Co-reporter:Mangesh Joshi;Carolyn Vargas;Prisca Boisguerin Dr.;Annette Diehl Dr.;Gerd Krause Dr.;Peter Schmieder Dr.;Karin Moelling Dr.;Volker Hagen Dr.;Markus Schade Dr. Dr.
Angewandte Chemie 2006 Volume 118(Issue 23) pp:
Publication Date(Web):3 MAY 2006
DOI:10.1002/ange.200503965
Tasche gefunden: Ob sich Protein-Protein-Wechselwirkungsdomänen als Angriffsstellen für Wirkstoffe eignen, ist noch umstritten. Die 3D-Struktur eines Komplexes aus einem kleinen organischen Liganden und der AF6-PDZ-Domäne lässt die Bildung einer Bindungstasche durch den Liganden erkennen (siehe Bild). Die Verbindung kann mit einem natürlichen Peptidliganden der Domäne konkurrieren und eignet sich als Baustein für die Erzeugung selektiver PDZ-Inhibitoren.
Co-reporter:Michele Fossi, Hartmut Oschkinat, Michael Nilges, Linda J. Ball
Journal of Magnetic Resonance 2005 Volume 175(Issue 1) pp:92-102
Publication Date(Web):July 2005
DOI:10.1016/j.jmr.2005.03.020
The calculation of protein structures from nuclear magnetic resonance (NMR) data has been greatly facilitated by improvements in software for the automatic assignment of NOESY spectra. Nevertheless, for larger proteins, resonance overlap may lead to an overwhelming number of assignment options per peak. Although most software for automatic NOESY assignment can deal with a certain level of assignment ambiguity, structure calculations fail when this becomes too high. Reducing the number of assignment options per peak by reducing the chemical shift tolerances can lead to correct assignments being excluded, and thus also to incorrect structures. We have investigated, systematically, for three proteins of different size, the influence of the chemical shift tolerance limits (Δ) and of the number of simulated annealing (SA) cooling steps on the performance of the software ARIA. Large tolerance windows, and the correspondingly high levels of ambiguity, did not cause problems when appropriately slower cooling was used in our SA protocol. In cases where a high percentage of well-converged structures was not achieved, we demonstrate that it is more productive to calculate fewer structures whilst applying slow cooling, than to calculate many structures with fast cooling. In this way, high-quality structures were obtained even for proteins whose NMR spectra showed great degeneracy, and where there was much inconsistency in peak alignment between different samples. The method described herein opens the way to the automated structure determination of larger proteins from NMR data.
Co-reporter:Michele Fossi;Federica Castellani Dr.;Michael Nilges Dr. Dr.;Barth-Jan van Rossum Dr.
Angewandte Chemie 2005 Volume 117(Issue 38) pp:
Publication Date(Web):20 SEP 2005
DOI:10.1002/ange.200501884
Der Flüssig-fest-Übergang: Automatische Zuordnung von Festkörper-NMR-spektroskopischen Daten sowie schnelle und genaue Berechnung der Struktur immobilisierter Proteine mit atomarer Auflösung gelingen mit dem Programm SOLARIA (einer modifizierten Version des ARIA-Protokolls). Im Bild sind die mit SOLARIA berechneten energetisch günstigsten Strukturen der SH3-Domäne von α-Spectrin (blau) mit dem Ergebnis einer Röntgenstrukturanalyse (rot) überlagert.
Co-reporter:Linda J. Ball Dr.;Ronald Kühne Dr.;Jens Schneider-Mergener Dr. Dr.
Angewandte Chemie 2005 Volume 117(Issue 19) pp:
Publication Date(Web):3 MAY 2005
DOI:10.1002/ange.200400618
Protein-Protein-Wechselwirkungen sind für alle Aspekte zellulärer Aktivität wichtig. Bildung und Dissoziation der Multiproteinkomplexe erfolgen auf spezifische Weise, oft mit ähnlichen Mechanismen. Bei der Signalübertragung sind häufig Proteindomänen beteiligt, die Prolin-reiche Motive binden (PRMs); Prolin begünstigt die spezifische Erkennung derartiger Motive, ohne dass dabei hohe Affinitäten benötigt werden. In diesem Aufsatz stellen wir eine detaillierte und quantitative Bewertung der Strukturmerkmale vor, die die Wechselwirkungen zwischen den PRM-Bindungsdomänen und den PRMs der Liganden festlegen, und untersuchen die Spezifität der PRM-Erkennung. Durch Kombination mit Ergebnissen des Screenings von Peptidbibliotheken konnten wir verschiedene hoch konservierte Wechselwirkungen identifizieren, die in allen Komplexen von PRM-Bindungsdomänen vorkommen. Das Unterdrücken von Protein-Protein-Wechselwirkungen mithilfe kleiner Moleküle ist eine große Herausforderung – daher ist es wichtig, zunächst genau die kritischen Wechselwirkungen zu identifizieren, die beim Design von Inhibitoren von PRM-Bindungsdomänen zu berücksichtigen sind.
Co-reporter:Linda J. Ball Dr.;Ronald Kühne Dr.;Jens Schneider-Mergener Dr. Dr.
Angewandte Chemie International Edition 2005 Volume 44(Issue 19) pp:
Publication Date(Web):3 MAY 2005
DOI:10.1002/anie.200400618
Protein–protein interactions are essential in every aspect of cellular activity. Multiprotein complexes form and dissociate constantly in a specifically tuned manner, often by conserved mechanisms. Protein domains that bind proline-rich motifs (PRMs) are frequently involved in signaling events. The unique properties of proline provide a mechanism for highly discriminatory recognition without requiring high affinities. We present herein a detailed, quantitative assessment of the structural features that define the interfaces between PRM-binding domains and their target PRMs, and investigate the specificity of PRM recognition. Together with the analysis of peptide-library screens, this approach has allowed the identification of several highly conserved key interactions found in all complexes of PRM-binding domains. The inhibition of protein–protein interactions by using small-molecule agents is very challenging. Therefore, it is important to first pinpoint the critical interactions that must be considered in the design of inhibitors of PRM-binding domains.
Co-reporter:Matthias Hiller Dipl.-Ing.;Ludwig Krabben Dr.;Kutti R. Vinothkumar;Federica Castellani Dr.;Barth-Jan van Rossum Dr.;Werner Kühlbrt and
ChemBioChem 2005 Volume 6(Issue 9) pp:
Publication Date(Web):1 SEP 2005
DOI:10.1002/cbic.200500132
Uniformly13C-,15N-labelled outer-membrane protein G (OmpG) from Escherichia coli was expressed for structural studies by solid-state magic-angle spinning (MAS) NMR. Inclusion bodies of the recombinant, labelled protein were purified under denaturing conditions and refolded in detergent. OmpG was reconstituted into lipid bilayers and several milligrams of two-dimensional crystals were obtained. Solid-state MAS NMR spectra showed signals with an apparent line width of 80–120 Hz (including homonuclear scalar couplings). Signal patterns for several amino acids, including threonines, prolines and serines were resolved and identified in 2D proton-driven spin-diffusion (PDSD) spectra.
Co-reporter:Michele Fossi, Federica Castellani, Michael Nilges, Hartmut Oschkinat,Barth-Jan van Rossum
Angewandte Chemie International Edition 2005 44(38) pp:6151-6154
Publication Date(Web):
DOI:10.1002/anie.200501884
Co-reporter:Federica Castellani;Barth van Rossum;Annette Diehl;Mario Schubert;Kristina Rehbein
Nature 2002 420(6911) pp:98-102
Publication Date(Web):2002-11-07
DOI:10.1038/nature01070
The determination of a representative set of protein structures is a chief aim in structural genomics. Solid-state NMR may have a crucial role in structural investigations of those proteins that do not easily form crystals or are not accessible to solution NMR, such as amyloid systems1 or membrane proteins2, 3, 4. Here we present a protein structure determined by solid-state magic-angle-spinning (MAS) NMR. Almost complete 13C and 15N resonance assignments for a micro-crystalline preparation of the α-spectrin Src-homology 3 (SH3) domain5 formed the basis for the extraction of a set of distance restraints. These restraints were derived from proton-driven spin diffusion (PDSD) spectra of biosynthetically site-directed, labelled samples obtained from bacteria grown using [1,3-13C]glycerol or [2-13C]glycerol as carbon sources. This allowed the observation of long-range distance correlations up to ~7 Å. The calculated global fold of the α-spectrin SH3 domain is based on 286 inter-residue 13C–13C and six 15N–15N restraints, all self-consistently obtained by solid-state MAS NMR. This MAS NMR procedure should be widely applicable to small membrane proteins that can be expressed in bacteria.
Co-reporter:Heiko Patzelt;Bernd Simon;Antonius terLaak;Brigitte Kessler;Ronald Kühne;Peter Schmieder;Dieter Oesterhelt
PNAS 2002 Volume 99 (Issue 15 ) pp:9765-9770
Publication Date(Web):2002-07-23
DOI:10.1073/pnas.132253899
The two forms of bacteriorhodopsin present in the dark-adapted state, containing either all-trans or 13-cis,15-syn retinal, were examined by using solution state NMR, and their structures were determined. Comparison of the all-trans and the 13-cis,15-syn forms shows a shift in position of about 0.25 Å within the pocket of the protein. Comparing this to the 13-cis,15-anti chromophore of the catalytic cycle M-intermediate structure, the 13-cis,15-syn form demonstrates a less pronounced up-tilt of the retinal C12—C14 region, while leaving W182 and T178 essentially unchanged.
The N—H dipole of the Schiff base orients toward the extracellular side in both forms, however, it reorients toward the intracellular
side in the 13-cis,15-anti configuration to form the catalytic M-intermediate. Thus, the change of the N—H dipole is considered primarily responsible
for energy storage, conformation changes of the protein, and the deprotonation of the Schiff base. The structural similarity
of the all-trans and 13-cis,15-syn forms is taken as strong evidence for the ion dipole dragging model by which proton (hydroxide ion) translocation follows
the change of the dipole.
Co-reporter:Barth-Jan van Rossum Dr.;Federica Castellani;Kristina Rehbein;Jutta Pauli Dr.
ChemBioChem 2001 Volume 2(Issue 12) pp:
Publication Date(Web):26 NOV 2001
DOI:10.1002/1439-7633(20011203)2:12<906::AID-CBIC906>3.0.CO;2-M
The assignment of nonexchanging protons of a small microcrystalline protein, the α-spectrin SH3 domain (7.2 kDa, 62 residues), was achieved by means of three-dimensional (3D) heteronuclear (1H–13C–13C) magic-angle spinning (MAS) NMR dipolar correlation spectroscopy. With the favorable combination of a high B0-field, a moderately high spinning frequency, and frequency-switched Lee-Goldburg irradiation applied during 1H evolution, a proton linewidth ≤0.5 ppm at 17.6 Tesla was achieved for the particular protein preparation used. A comparison of the solid-state 1H chemical shifts with the shifts found in solution shows a remarkable similarity, which reflects the identical protein structures in solution and in the solid. Significant differences between the MAS solid- and liquid-state 1H chemical shifts are only observed for residues that are located at the surface of the protein and that exhibit contacts between different SH3 molecules. In two cases, aromatic residues of neighboring SH3 molecules induce pronounced upfield ring-current shifts for protons in the contact area.
Co-reporter:Jutta Pauli Dr.;Marc Baldus Dr.;Barth van Rossum Dr.;Huub de Groot
ChemBioChem 2001 Volume 2(Issue 4) pp:
Publication Date(Web):23 MAR 2001
DOI:10.1002/1439-7633(20010401)2:4<272::AID-CBIC272>3.0.CO;2-2
The backbone and side-chain 13C and 15N signals of a solid 62-residue (u-13C,15N)-labelled protein containing the α-spectrin SH3 domain were assigned by two-dimensional (2D) magic angle spinning (MAS) 15N–13C and 13C–13C dipolar correlation spectroscopy at 17.6 T. The side-chain signal sets of the individual amino acids were identified by 2D 13C–13C proton-driven spin diffusion and dipolar recoupling experiments. Correlations to the respective backbone nitrogen signals were established by 2D NCACX (CX=any carbon atom) experiments, which contain a proton–nitrogen and a nitrogen–carbon cross-polarisation step followed by a carbon–carbon homonuclear transfer unit. Interresidue correlations leading to sequence-specific assignments were obtained from 2D NCOCX experiments. The assignment is nearly complete for the SH3 domain residues 7–61, while the signals of the N- and C-terminal residues 1–6 and 62, respectively, outside the domain boundaries are not detected in our MAS spectra. The resolution observed in these spectra raises expectations that receptor-bound protein ligands and slightly larger proteins (up to 20 kDa) can be readily assigned in the near future by using three-dimensional versions of the applied or analogous techniques.
Co-reporter:Mark J. S. Kelly;Linda J. Ball;Cornelia Krieger;Yihua Yu;Markus Fischer;Susanne Schiffmann;Peter Schmieder;Ronald Kühne;Wolfgang Bermel;Adelbert Bacher;Gerald Richter
PNAS 2001 Volume 98 (Issue 23 ) pp:13025-13030
Publication Date(Web):2001-11-06
DOI:10.1073/pnas.231323598
Recent developments in NMR have extended the size range of proteins amenable to structural and functional characterization
to include many larger proteins involved in important cellular processes. By applying a combination of residue-specific isotope
labeling and protein deuteration strategies tailored to yield specific information, we were able to determine the solution
structure and study structure–activity relationships of 3,4-dihydroxy-2-butanone-4-phosphate synthase, a 47-kDa enzyme from
the Escherichia coli riboflavin biosynthesis pathway and an attractive target for novel antibiotics. Our investigations of the enzyme's ligand
binding by NMR and site-directed mutagenesis yields a conclusive picture of the location and identity of residues directly
involved in substrate binding and catalysis. Our studies illustrate the power of state-of-the-art NMR techniques for the structural
characterization and investigation of ligand binding in protein complexes approaching the 50-kDa range in solution.
Co-reporter:Helena Domingues;David Cregut;Walter Sebald;Luis Serrano
Nature Structural and Molecular Biology 1999 6(7) pp:652-656
Publication Date(Web):1999-07-01
DOI:10.1038/10706
In this work we describe the rational design of two helix coiled coil peptide
mimetics of interleukin-4 (IL-4) which are able to recognize and bind its
high affinity receptor (IL-4R). We have used the leucine-zipper domain
of the yeast transcription factor GCN4 as a scaffold into which the putative
binding epitope of IL-4 for IL-4R was transferred in a stepwise manner,
using computer-aided molecular modeling. The resulting molecules bind IL-4R
with affinities ranging from 2 mM to 5 M, depending on the fraction of
the IL-4 binding site incorporated and on their stability. To our knowledge
this is the first time a molecule capable of binding a cytokine receptor has
been successfully designed in a rational manner.
Co-reporter:
Nature Structural and Molecular Biology 1999 6(5) pp:408-410
Publication Date(Web):
DOI:10.1038/8203
A new role for certain PDZ domains has been revealed — binding to
non-terminal -hairpin structures in other proteins. These interactions
involve the same binding site as is used for binding to the canonical C-terminal
peptide targets of PDZ domains.
Co-reporter:W. Trent Franks, Arne H. Linden, Britta Kunert, Barth-Jan van Rossum, Hartmut Oschkinat
European Journal of Cell Biology (April 2012) Volume 91(Issue 4) pp:340-348
Publication Date(Web):1 April 2012
DOI:10.1016/j.ejcb.2011.09.002
Structural biology is developing into a universal tool for visualizing biological processes in space and time at atomic resolution. The field has been built by established methodology like X-ray crystallography, electron microscopy and solution NMR and is now incorporating new techniques, such as small-angle X-ray scattering, electron tomography, magic-angle-spinning solid-state NMR and femtosecond X-ray protein nanocrystallography. These new techniques all seek to investigate non-crystalline, native-like biological material. Solid-state NMR is a relatively young technique that has just proven its capabilities for de novo structure determination of model proteins. Further developments promise great potential for investigations on functional biological systems such as membrane-integrated receptors and channels, and macromolecular complexes attached to cytoskeletal proteins. Here, we review the development and applications of solid-state NMR from the first proof-of-principle investigations to mature structure determination projects, including membrane proteins. We describe the development of the methodology by looking at examples in detail and provide an outlook towards future ‘big’ projects.
Co-reporter:Christian Köhler, Janet K. Lighthouse, Tobias Werther, Olav M. Andersen, ... Hartmut Oschkinat
Structure (9 March 2011) Volume 19(Issue 3) pp:337-348
Publication Date(Web):9 March 2011
DOI:10.1016/j.str.2010.12.022
Mesoderm development (MESD) is a 224 amino acid mouse protein that acts as a molecular chaperone for the low-density lipoprotein receptor (LDLR) family. Here, we provide evidence that the region 45–184 of MESD is essential and sufficient for this function and suggest a model for its mode of action. NMR studies reveal a β−α−β−β−α−β core domain with an α-helical N-terminal extension that interacts with the β sheet in a dynamic manner. As a result, the structural ensemble contains open (active) and closed (inactive) forms, allowing for regulation of chaperone activity through substrate binding. The mutant W61R, which is lethal in Drosophila, adopts only the open state. The receptor motif recognized by MESD was identified by in vitro-binding studies. Furthermore, in vivo functional evidence for the relevance of the identified contact sites in MESD is provided.Highlights► Solution NMR structure of the conserved part of MESD ► Measurements of internal dynamics reveal a two-state conformer ► Identification of MESD's contact site to LRP's by in vitro and in vivo data ► Suggestion of a mechanism for the MESD-assisted folding of LDLR family members
Co-reporter:Stefan Jehle, Barth van Rossum, Joseph R. Stout, Satoshi M. Noguchi, ... Ponni Rajagopal
Journal of Molecular Biology (6 February 2009) Volume 385(Issue 5) pp:1481-1497
Publication Date(Web):6 February 2009
DOI:10.1016/j.jmb.2008.10.097
Atomic-level structural information on αB-Crystallin (αB), a prominent member of the small heat-shock protein family, has been a challenge to obtain due its polydisperse oligomeric nature. We show that magic-angle spinning solid-state NMR can be used to obtain high-resolution information on an ∼ 580-kDa human αB assembled from 175-residue 20-kDa subunits. An ∼ 100-residue α-crystallin domain is common to all small heat-shock proteins, and solution-state NMR was performed on two different α-crystallin domain constructs isolated from αB. In vitro, the chaperone-like activities of full-length αB and the isolated α-crystallin domain are identical. Chemical shifts of the backbone and Cβ resonances have been obtained for residues 64–162 (α-crystallin domain plus part of the C-terminus) in αB and the isolated α-crystallin domain by solid-state and solution-state NMR, respectively. Both sets of data strongly predict six β-strands in the α-crystallin domain. A majority of residues in the α-crystallin domain have similar chemical shifts in both solid-state and solution-state, indicating similar structures for the domain in its isolated and oligomeric forms. Sites of intersubunit interaction are identified from chemical shift differences that cluster to specific regions of the α-crystallin domain. Multiple signals are observed for the resonances of M68 in the oligomer, identifying the region containing this residue as existing in heterogeneous environments within αB. Evidence for a novel dimerization motif in the human α-crystallin domain is obtained by a comparison of (i) solid-state and solution-state chemical shift data and (ii) 1H–15N heteronuclear single quantum coherence spectra as a function of pH. The isolated α-crystallin domain undergoes a dimer–monomer transition over the pH range 7.5–6.8. This steep pH-dependent switch may be important for αB to function optimally (e.g., to preserve the filament integrity of cardiac muscle proteins such as actin and desmin during cardiac ischemia, which is accompanied by acidosis).
Co-reporter:Ludwig Krabben, Barth-Jan van Rossum, Stefan Jehle, Eduard Bocharov, ... Hartmut Oschkinat
Journal of Molecular Biology (24 July 2009) Volume 390(Issue 4) pp:662-671
Publication Date(Web):24 July 2009
DOI:10.1016/j.jmb.2009.05.016
The contact area of neurotoxin II from Naja naja oxiana when interacting with the membrane-bound nicotinic acetylcholine receptor from Torpedo californica was determined by solid-state, magic-angle spinning NMR spectroscopy. For this purpose, the carbon signals for more than 90% of the residues of the bound neurotoxin were assigned. Differences between the solution and solid-state chemical shifts of the free and bound form of the toxin are confined to distinct surface regions. Loop II of the short toxin was identified as the main interaction site. In addition, loop III of neurotoxin II shows several strong responses defining an additional interaction site. A comparison with the structures of α-cobratoxin bound to the acetylcholine-binding protein from snail species Lymnaea stagnalis and Aplysia californica, and of α-bungarotoxin bound to an extracellular domain of an α-subunit of the receptor reveals different contact areas for long and short α-neurotoxins.
Co-reporter:Michel-Andreas Geiger, Marcella Orwick-Rydmark, Katharina Märker, W. Trent Franks, Dmitry Akhmetzyanov, Daniel Stöppler, Maximilian Zinke, Edgar Specker, Marc Nazaré, Anne Diehl, Barth-Jan van Rossum, Fabien Aussenac, Thomas Prisner, Ümit Akbey and Hartmut Oschkinat
Physical Chemistry Chemical Physics 2016 - vol. 18(Issue 44) pp:NaN30704-30704
Publication Date(Web):2016/10/28
DOI:10.1039/C6CP06154K
Dynamic nuclear polarization exploits electron spin polarization to boost signal-to-noise in magic-angle-spinning (MAS) NMR, creating new opportunities in materials science, structural biology, and metabolomics studies. Since protein NMR spectra recorded under DNP conditions can show improved spectral resolution at 180–200 K compared to 110 K, we investigate the effects of AMUPol and various deuterated TOTAPOL isotopologues on sensitivity and spectral resolution at these temperatures, using proline and reproducibly prepared SH3 domain samples. The TOTAPOL deuteration pattern is optimized for protein DNP MAS NMR, and signal-to-noise per unit time measurements demonstrate the high value of TOTAPOL isotopologues for Protein DNP MAS NMR at 180–200 K. The combined effects of enhancement, depolarization, and proton longitudinal relaxation are surprisingly sample-specific. At 200 K, DNP on SH3 domain standard samples yields a 15-fold increase in signal-to-noise over a sample without radicals. 2D and 3D NCACX/NCOCX spectra were recorded at 200 K within 1 and 13 hours, respectively. Decreasing enhancements with increasing 2H-content at the CH2 sites of the TEMPO rings in CD3-TOTAPOL highlight the importance of protons in a sphere of 4–6 Å around the nitroxyl group, presumably for polarization pickup from electron spins.
Co-reporter:Anil P. Jagtap, Michel-Andreas Geiger, Daniel Stöppler, Marcella Orwick-Rydmark, Hartmut Oschkinat and Snorri Th. Sigurdsson
Chemical Communications 2016 - vol. 52(Issue 43) pp:NaN7023-7023
Publication Date(Web):2016/05/10
DOI:10.1039/C6CC01813K
Dynamic nuclear polarization (DNP) is an efficient method to overcome the inherent low sensitivity of magic-angle spinning (MAS) solid-state NMR. We report a new polarizing agent (bcTol), designed for biological applications, that yielded an enhancement value of 244 in a microcrystalline SH3 domain sample at 110 K.