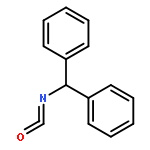
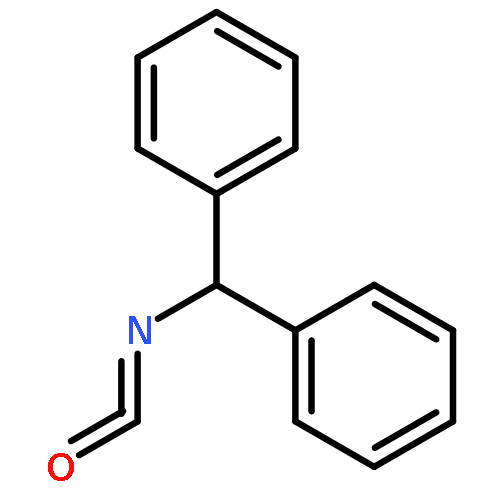
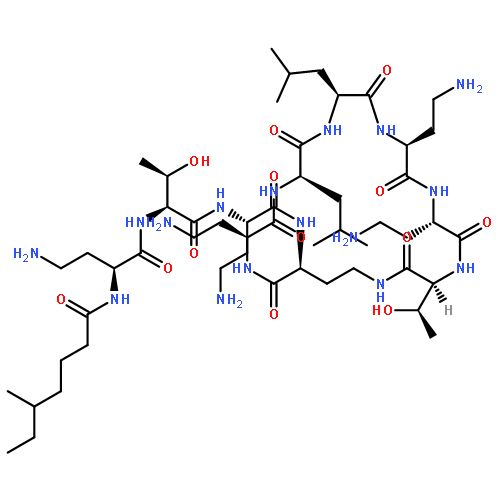
Co-reporter:Bo Wang; Boobalan Pachaiyappan; Jordon D. Gruber; Michael G. Schmidt; Yong-Mei Zhang;Patrick M. Woster
Journal of Medicinal Chemistry 2016 Volume 59(Issue 7) pp:3140-3151
Publication Date(Web):March 10, 2016
DOI:10.1021/acs.jmedchem.5b01912
Antibiotic resistance is a growing threat to human health exacerbated by a lack of new antibiotics. We now describe a series of substituted diamines that produce rapid bactericidal activity against both Gram-positive and Gram-negative bacteria, including methicillin-resistant Staphylococcus aureus and stationary-phase bacteria. These compounds reduce biofilm formation and promote biofilm dispersal in Pseudomonas aeruginosa. The most potent analogue, 3 (1,13-bis{[(2,2-diphenyl)-1-ethyl]thioureido}-4,10-diazatridecane), primarily acts by depolarization of the cytoplasmic membrane and permeabilization of the bacterial outer membrane. Transmission electron microscopy confirmed that 3 disrupts membrane integrity rapidly. Compound 3 is also synergistic with kanamycin, demonstrated by the checkerboard method and by time-kill kinetic experiments. In human cell toxicity assays, 3 showed limited adverse effects against the HEK293T human kidney embryonic cells and A549 human adenocarcinoma cells. In addition, 3 produced no adverse effects on Caenorhabditis elegans development, survival, and reproduction. Collectively, diamines related to 3 represent a new class of broad-spectrum antibacterials against drug-resistant pathogens.
Co-reporter:Youxuan Li and Patrick M. Woster
MedChemComm 2015 vol. 6(Issue 4) pp:613-618
Publication Date(Web):16 Dec 2014
DOI:10.1039/C4MD00401A
Small molecules featuring a hydroxamic acid or a benzamide zinc binding group (ZBG) are the most thoroughly studied histone deacetylase (HDAC) inhibitors. However, concerns about the pharmacokinetic liabilities of the hydroxamic acid moiety and potential metabolic toxicity of the aniline portion of benzamideHDAC inhibitors have stimulated research efforts aimed at discovering alternative ZBGs. Here we report the 2-(oxazol-2-yl)phenol moiety as a novel ZBG that can be used to produce compounds that are potent HDAC inhibitors. A series of analogues with this novel ZBG have been synthesized, and these analogues exhibit selective inhibition against HDAC1 as well as the class IIb HDACs (HDAC6 and HDAC10). Compound 10 possesses an IC50 value of 7.5 μM in the MV-4-11 leukemia cell line, and 2.5 μM compound 10 induces a comparable amount of acetylated histone 3 lysine 9 (H3K9) and p21Waf1/CIP1 as 0.5 μM of SAHA. Modeling of compound 10 in the active site of HDAC2 demonstrates that the 2-(oxazol-2-yl)phenol moiety has a zinc-binding pattern similar to benzamideHDAC inhibitors.
Co-reporter:Shannon L. Nowotarski, Boobalan Pachaiyappan, Steven L. Holshouser, Craig J. Kutz, Youxuan Li, Yi Huang, Shiv K. Sharma, Robert A. Casero Jr., Patrick M. Woster
Bioorganic & Medicinal Chemistry 2015 23(7) pp: 1601-1612
Publication Date(Web):
DOI:10.1016/j.bmc.2015.01.049
Co-reporter:Isuru R. Kumarasinghe and Patrick M. Woster
ACS Medicinal Chemistry Letters 2014 Volume 5(Issue 1) pp:29-33
Publication Date(Web):November 8, 2013
DOI:10.1021/ml4002997
Lysine specific demethylase 1 (LSD1) selectively removes methyl groups from mono- and dimethylated histone 3 lysine 4 (H3K4), resulting in gene silencing. LSD1 is overexpressed in many human cancers, resulting in aberrant silencing of tumor suppressor genes. Thus, LSD1 is a validated target for the discovery of antitumor agents. Using a ligand-based approach, we designed and synthesized a series of cyclic and linear peptides that are effective inhibitors of LSD1. Linear peptide 7 and cyclic peptide 9 inhibited LSD1 in vitro by 91 and 94%, respectively, at a concentration of 10 μM. Compound 9 was a potent LSD1 inhibitor (IC50 2.1 μM; Ki 385 nM) and had moderate antitumor activity in the MCF-7 and Calu-6 cell lines in vitro. Importantly, 9 is significantly more stable to hydrolysis in rat plasma than the linear analogue 7. The cyclic peptides described herein represent important lead structures in the search for inhibitors of flavin-dependent histone demethylases.Keywords: Chromatin architecture; chromatin remodeling; cyclic peptide; histone; histone 3 lysine 4; KDM inhibitors; lysine specific demethylase;
Co-reporter:Boobalan Pachaiyappan, Patrick M. Woster
Bioorganic & Medicinal Chemistry Letters 2014 Volume 24(Issue 1) pp:21-32
Publication Date(Web):1 January 2014
DOI:10.1016/j.bmcl.2013.11.001
The field of epigenetics has expanded rapidly to reveal multiple new targets for drug discovery. The functional elements of the epigenomic machinery can be categorized as writers, erasers and readers, and together these elements control cellular gene expression and homeostasis. It is increasingly clear that aberrations in the epigenome can underly a variety of diseases, and thus discovery of small molecules that modulate the epigenome in a specific manner is a viable approach to the discovery of new therapeutic agents. In this Digest, the components of epigenetic control of gene expression will be briefly summarized, and efforts to identify small molecules that modulate epigenetic processes will be described.Figure optionsDownload full-size imageDownload high-quality image (161 K)Download as PowerPoint slide
Co-reporter:Craig J. Kutz, Steven L. Holshouser, Ethan A. Marrow and Patrick M. Woster
MedChemComm 2014 vol. 5(Issue 12) pp:1863-1870
Publication Date(Web):22 Aug 2014
DOI:10.1039/C4MD00283K
The chromatin remodeling amine oxidase lysine-specific demethylase 1 (LSD1) has become an attractive target for the design of specific inhibitors with therapeutic potential. We, and others, have described LSD1 inhibitors that have potential as antitumor agents. Many of the currently known LSD1 inhibitors are poor drug candidates, or are structurally based on the tranylcypromine backbone, thus increasing the potential for off-target effects mediated by other amine oxidases. We now describe a series of potent LSD1 inhibitors based on a novel 1,2,4-triazole scaffold; these inhibitors show a high degree of specificity for LSD1 in vitro, and cause increases in cellular histone 3 dimethyllysine 4 (H3K4me2), a gene transcription activating mark. Importantly, these inhibitors exhibit low toxicity towards mammalian cells in vitro, and thus they may show utility in the treatment of epigenetically-based diseases where cell death is not a desired endpoint.
Co-reporter:Stuart Hazeldine ; Boobalan Pachaiyappan ; Nora Steinbergs ; Shannon Nowotarski ; Allison S. Hanson ; Robert A. Casero ; Jr.;Patrick M. Woster
Journal of Medicinal Chemistry 2012 Volume 55(Issue 17) pp:7378-7391
Publication Date(Web):August 9, 2012
DOI:10.1021/jm3002845
The recently discovered enzyme lysine-specific demethylase 1 (LSD1) plays an important role in the epigenetic control of gene expression, and aberrant gene silencing secondary to LSD1 dysregulation is thought to contribute to the development of cancer. We reported that (bis)guanidines, (bis)biguanides, and their urea- and thiourea isosteres are potent inhibitors of LSD1 and induce the re-expression of aberrantly silenced tumor suppressor genes in tumor cells in vitro. We now report a series of small molecule amidoximes that are moderate inhibitors of recombinant LSD1 but that produce dramatic changes in methylation at the histone 3 lysine 4 (H3K4) chromatin mark, a specific target of LSD1, in Calu-6 lung carcinoma cells. In addition, these analogues increase cellular levels of secreted frizzle-related protein (SFRP) 2, H-cadherin (HCAD), and the transcription factor GATA4. These compounds represent leads for an important new series of drug-like epigenetic modulators with the potential for use as antitumor agents.
Co-reporter:Shiv K. Sharma, Stuart Hazeldine, Michael L. Crowley, Allison Hanson, Ross Beattie, Sheeba Varghese, Thulani M. D. Senanayake, Aiko Hirata, Fusao Hirata, Yi Huang, Yu Wu, Nora Steinbergs, Tracey Murray-Stewart, Ian Bytheway, Robert A. Casero and Patrick M. Woster
MedChemComm 2012 vol. 3(Issue 1) pp:14-21
Publication Date(Web):25 Oct 2011
DOI:10.1039/C1MD00220A
Chromatin remodelling enzymes such as the histone deacetylases (HDACs) and histone demethylases such as lysine-specific demethylase 1 (LSD1) have been validated as targets for cancer drug discovery. Although a number of HDACinhibitors have been marketed or are in human clinical trials, the search for isoform-specific HDACinhibitors is an ongoing effort. In addition, the discovery and development of compounds targeting histone demethylases are in their early stages. Epigenetic modulators used in combination with traditional antitumor agents such as 5-azacytidine represent an exciting new approach to cancer chemotherapy. We have developed multiple series of HDACinhibitors and LSD1inhibitors that promote the re-expression of aberrantly silenced genes that are important in human cancer. The design, synthesis and biological activity of these analogues is described herein.







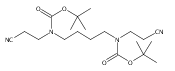
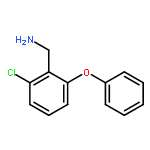
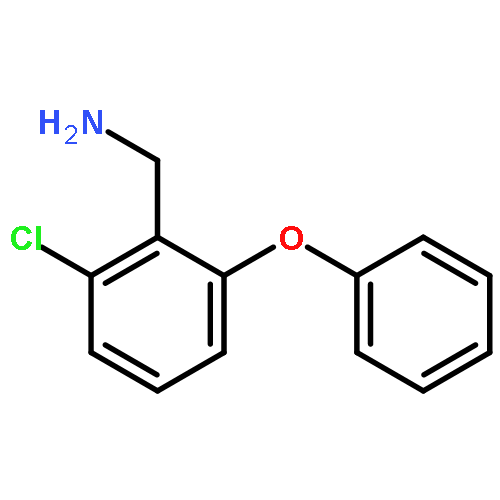


![1H-1,2,4-Triazol-3-amine, N-[(2,6-dichlorophenyl)methyl]-](http://img.cochemist.com/ccimg/90700/90626-44-1.png)
![1H-1,2,4-Triazol-3-amine, N-[(2,6-dichlorophenyl)methyl]-](http://img.cochemist.com/ccimg/90700/90626-44-1_b.png)