Co-reporter:Juliane Schmid, Wolfgang Frey, and René Peters
Organometallics November 13, 2017 Volume 36(Issue 21) pp:4313-4313
Publication Date(Web):October 31, 2017
DOI:10.1021/acs.organomet.7b00729
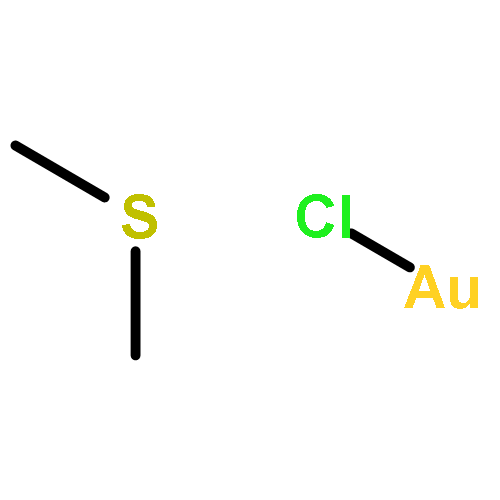
Salen ligands and mesoionic carbene (MIC) ligands constitute important ligand classes in homogeneous catalysis. In this article we describe the first integration of a salen moiety and MIC donors in the same catalyst entity. Homo- and heterometallic polynuclear metal complexes could be selectively prepared by first installing a metal center (Ni(II), Pd(II)) in the N2O2 salen sphere followed by metalation of the mesoionic carbenes (Ag(I), Pd(II)). The silver complexes could be used for transmetalations to form Au(III) complexes. Several complexes showed catalytic activity in the 1,4-addition of an oxindole to a nitroolefin.
Co-reporter:Florian Broghammer;Daniel Brodbeck;Thorsten Junge;René Peters
Chemical Communications 2017 vol. 53(Issue 6) pp:1156-1159
Publication Date(Web):2017/01/17
DOI:10.1039/C6CC09774J
Epoxide desymmetrizations by bromide are very rare despite the large synthetic potential of chiral bromohydrins. Herein we present a new concept for epoxide desymmetrizations in which a bifunctional Lewis acid/ammonium salt catalyst allows for efficient enantioselective epoxide ring openings by Br−. With acetylbromide as a Br− source bromohydrin esters are formed.
Co-reporter:
Chemistry - A European Journal 2017 Volume 23(Issue 10) pp:2448-2460
Publication Date(Web):2017/02/16
DOI:10.1002/chem.201605244
AbstractBenzylic N-substituted stereocenters constitute a frequent structural motif in drugs. Their highly enantioselective generation is hence of technical importance. An attractive strategy is the arylation of imines with organoboron reagents. Chiral Rh complexes have reached a high level of productivity for this reaction type. In this article we describe that an electron rich PdII catalyst also performs well in the arylation of aldimines, comparable to the best Rh catalysts. The ferrocenyl palladacycle-acetate catalyst allows for a broad substrate scope and very high enantioselectivities. Commonly observed side reactions like aryl–aryl homocouplings and imine hydrolysis could be blocked. Mechanistic studies implicate that a) the acetate ligand is crucial for transmetallation, b) the active catalyst is most likely a palladacycle-OAc monomer, c) the rate limiting step is probably the product release. By added KOAc the arylation could also be applied to ketimines.
Co-reporter:Katja Latendorf;Melanie Mechler;Irina Schamne;Daniel Mack;Wolfgang Frey;René Peters
European Journal of Organic Chemistry 2017 Volume 2017(Issue 28) pp:4140-4167
Publication Date(Web):2017/08/02
DOI:10.1002/ejoc.201700436
Lewis acid catalysis and nucleophilic carbene catalysis are complementary fundamental concepts to accelerate and control chemical reactions of aldehyde substrates. Their efficient merger has recently been achieved using two separate catalyst species. The present report describes our efforts to develop a cooperative catalyst system which incorporates both features – Lewis acid and nucleophilic NHC – within the same catalyst entity. To generate free carbene moieties under very mild conditions, Ag-NHC complexes were formed releasing the nucleophilic carbene upon treatment with PPh3. The result is the formation of an enol-δ-lactone as new enal dimerization product. Silver is essential for the reactivity mode thus suggesting that it plays a double role in the catalytic event.
Co-reporter:Marcel Weiss;Julia Holz ;René Peters
European Journal of Organic Chemistry 2016 Volume 2016( Issue 1) pp:210-227
Publication Date(Web):
DOI:10.1002/ejoc.201501290
Abstract
Allylic substitution reactions provide a valuable tool for the functionalization of CH acidic pronucleophiles. Often, control over the stereocenter generated at the nucleophilic reactant is still a challenge. The majority of studies that address this issue employ metal complexes with a low metal oxidation state (e.g. Pd0) to form allyl complexes through oxidative addition. In this article we describe the use of heterobimetallic PtII/PdII complexes, which probably activate the olefinic substrates through an SN2′ pathway. The reaction of α-cyanoacetates delivers linear allylation products with exclusive regioselectivity and high E/Z-selectivity for the new C=C double bond. Although the enantioselectivities attained are moderate, they are significantly higher than with related mono-PdII or -PtII catalysts or the corresponding bis-PdII complex, which indicates cooperation of the different metals. Control experiments suggest simultaneous activation of both reaction partners.
Co-reporter:Dr. Johannes Moritz Bauer;Dr. Wolfgang Frey ;Dr. René Peters
Chemistry - A European Journal 2016 Volume 22( Issue 16) pp:5767-5777
Publication Date(Web):
DOI:10.1002/chem.201600138
Abstract
The streamlined catalytic access to enantiopure allylic amines as valuable precursors towards chiral β- and γ-aminoalcohols as well as α- and β-aminoacids is desirable for industrial purposes. In this article an enantioselective method is described that transforms achiral allylic alcohols and N-tosylisocyanate in a single step into highly enantioenriched N-tosyl protected allylic amines via an allylic carbamate intermediate. The latter is likely to undergo a cyclisation-induced [3,3]-rearrangement catalysed by a planar chiral pentaphenylferrocene palladacycle in cooperation with a tertiary amine base. The otherwise often indispensable activation of palladacycle catalysts by a silver salt is not required in the present case and there is also no need for an inert gas atmosphere. To further improve the synthetic value, the rearrangement was used to form dimethylaminosulfonyl-protected allylic amines, which can be deprotected under non-reductive conditions.
Co-reporter:Marcel Weiss and René Peters
ACS Catalysis 2015 Volume 5(Issue 1) pp:310
Publication Date(Web):December 8, 2014
DOI:10.1021/cs501495g
Transition-metal-catalyzed cross-coupling reactions between sp2-hybridized C atoms are of prime importance in both target and diversity oriented synthesis. Ideal cross-coupling reactions would neither require any leaving groups nor stoichiometric reagents. In this article, we report the first direct dehydrogenative cross-couplings between aromatic C–H bonds (in most cases using indole substrates) and allylic alcohols, which do not require an additional classical stoichiometric oxidizing agent and provide β-arylketones as value-added products. Ruthenocene- or ferrocene-based bismetallacycles, in which either Pd(II) or Pt(II) are the catalytically active centers, were found to be particularly efficient catalysts. Control experiments suggest that the bismetallacycles initially transform the allylic alcohols into vinylketones, which then alkylate the aromatic substrate in the presence of the catalyst. The fact that the dehydrogenative coupling does not require a classical stoichiometric oxidizing agent is explained either by protonolysis of a metallacyclic M(II)-H intermediate or by a mechanism in which an excess of the allylic alcohol substrate serves as a sacrificial hydrogen acceptor. The title reaction is supported by cocatalytic amounts of Ni(OAc)2. In preliminary studies, it was observed that the title reaction can as well be applied to prochiral CH-acidic pronucleophiles such as α-cyanoacetates, representing the first examples for direct enantioselective β-ketoalkylations via allylic alcohols in the absence of an additional oxidant.Keywords: bimetallic catalysts; bispalladacycles; dehydrogenative cross-coupling; direct cross-coupling; ferrocene; indole; oxidant-free; platinacycle; ruthenocene; α-cyanoacetate; β-ketoalkylations
Co-reporter:Marta Ayerbe Garcia, Wolfgang Frey, Mark R. Ringenberg, Max Schwilk and René Peters
Chemical Communications 2015 vol. 51(Issue 94) pp:16806-16809
Publication Date(Web):05 Oct 2015
DOI:10.1039/C5CC07018J
Oxidation of Au(I) in the presence of Fe(II) allowed for the synthesis of unique dinuclear ferrocenyl Au(II) complexes via the first reported enantiopure planar chiral ferrocenyl Au(I) complex. (Spectro)electrochemical studies show that oxidation at Fe(II) is favoured, but DFT studies suggest that the energy differences for oxidation of one or the other metal should be quite small.
Co-reporter:J. Moritz Bauer and René Peters
Catalysis Science & Technology 2015 vol. 5(Issue 4) pp:2340-2346
Publication Date(Web):06 Feb 2015
DOI:10.1039/C4CY01749H
A streamlined synthetic access to enantiomerically pure allylic amines as advanced and valuable synthetic building blocks is important for pharmaceutical sciences. The rearrangement of allylic trihaloacetimidates is known as an attractive method to furnish branched chiral allylic trihaloacetamides with high levels of enantio- and regioselectivity. In the present article we report our studies on the catalytic asymmetric rearrangement of the corresponding non-halogenated acetimidates, which might provide economic advantages by avoiding CX3 groups. The regioselective title reaction proceeds with high levels of enantioselectivity, provides high yields and requires only low catalyst loadings. In addition, the generated N-acetyl and N-phenacetyl functionalities offer the option of a subsequent mild enzymatic amide hydrolysis to get access to nearly enantiopure allylic amines.
Co-reporter:Dipl.-Chem. Tina Hellmuth;Dr. Wolfgang Frey ;Dr. René Peters
Angewandte Chemie International Edition 2015 Volume 54( Issue 9) pp:2788-2791
Publication Date(Web):
DOI:10.1002/anie.201410933
Abstract
Isoxazolinones constitute a class of heterocycles utilized for the development of novel drug candidates. The cyclic oxime ester motif is also synthetically useful as it contains functional handles which have previously been used to provide access to an assortment of valuable compound classes not easily accessible by alternative approaches. However, asymmetric methods towards isoxazolinones are notoriously scarce. Herein we report the first catalytic asymmetric alkylations of isoxazolinones forming all-C-substituted quaternary stereocenters. The present studies were driven by the question of how to control the regioselectivity in the competition of different nucleophilic positions. The investigation of a direct 1,4-addition uncovered that a sterically demanding palladacycle catalyst directs the reactivity in the absence of a base nearly exclusively to the nucleophilic C atom, while at the same time it allows for high enantioselectivity and TONs up to 1900.
Co-reporter:Stefan Rieckhoff, Tina Hellmuth, and René Peters
The Journal of Organic Chemistry 2015 Volume 80(Issue 13) pp:6822-6830
Publication Date(Web):June 23, 2015
DOI:10.1021/acs.joc.5b01065
Substituted pyridines are prevalent heterocycles of fundamental importance. Their efficient regioselective preparation is often still a challenge despite a large number of reported synthetic methodologies. In this letter we report an operationally simple approach that makes use of readily accessible isoxazolinones. The protocol involves a Pd(II)-catalyzed C-regioselective 1,4-addition to vinylketones, followed by a Pd(0)-catalyzed transformation, which is assumed to proceed via vinylnitrene-Pd intermediates. Both hydrogen and air are necessary for the pyridine formation step and could be employed at ratios above the upper explosive limit thus avoiding a safety issue. This new strategy allows an effective, scalable and practical access to various previously unknown 2,3,6-trisubstituted pyridines.
Co-reporter:Dipl.-Chem. Carmen Schrapel ;Dr. René Peters
Angewandte Chemie 2015 Volume 127( Issue 35) pp:10428-10432
Publication Date(Web):
DOI:10.1002/ange.201501846
Abstract
Enantiomerenreine Benzylamine sind für die Wirkstoffentwicklung von großer Bedeutung. Ein leicht zugänglicher, planar-chiraler, Ferrocen-basierter Palladacyclus erwies sich als sehr effizienter enantioselektiver Katalysator zur Bildung N-substituierter benzylischer Stereozentren durch Beschleunigung der 1,2-Addition von Arylboroxinen an aromatische und aliphatische Imine. Für Aldimine war kein Einsatz einer exogenen Base zur Aktivierung des Boroxins erforderlich, wenn Acetat als anionischer Ligand verwendet wurde. Verbreitete Probleme, wie Aryl-Aryl-Homokupplungen und die Imin-Hydrolyse, konnten vollständig vermieden werden, letztere sogar ohne Einsatz von Molekularsieb.
Co-reporter:Dipl.-Chem. Melanie Mechler ;Dr. René Peters
Angewandte Chemie 2015 Volume 127( Issue 35) pp:10442-10446
Publication Date(Web):
DOI:10.1002/ange.201502930
Abstract
Diastereodivergenz ist eine Herausforderung für die katalytische asymmetrische Synthese. Für viele Reaktionstypen ist die Bildung eines Diastereomers inhärent bevorzugt, während das andere Diastereomer nicht direkt und effizient zugänglich ist, sondern umständliche Syntheseumwege erfordert. Das Umgehen dieser natürlichen Präferenz mithilfe eines Katalysators erfordert die räumliche Kontrolle über beide Reaktionspartner. Wir stellen einen neuartigen, polyfunktionellen Katalysator vor, in dem eine NiII-Bis(phenoxyimin)-Einheit, freie Hydroxygruppen und ein axial-chiraler Bisimidazolium-Baustein an der Stereokontrolle der direkten 1,4-Addition von Oxindolen an Nitroolefine beteiligt sind. Beide Epimere des 1,4-Adduktes sind nach Belieben durch Variation der Konstitution und Konfiguration des Liganden zugänglich. Wie bereits berichtet wurde, sind die Produkte wertvolle Vorstufen für Indolalkaloide, und die vorliegende Methode sollte somit den Zugang zu den epimeren Derivaten eröffnen.
Co-reporter:Dipl.-Chem. Carmen Schrapel ;Dr. René Peters
Angewandte Chemie International Edition 2015 Volume 54( Issue 35) pp:10289-10293
Publication Date(Web):
DOI:10.1002/anie.201501846
Abstract
Enantiomerically pure benzylic amines are important for the development of new drugs. A readily accessible planar-chiral ferrocene-derived palladacycle is shown to be a highly efficient catalyst for the formation of N-substituted benzylic stereocenters; this catalyst accelerates the 1,2-addition of arylboroxines to aromatic and aliphatic imines with exceptional levels of enantioselectivity. Using aldimines an exogenous base was not necessary for the activation of the boroxines, when acetate was used as an anionic ligand. Common problems such as aryl–aryl homocouplings and imine hydrolysis were fully overcome, the latter even in the absence of molecular sieves.
Co-reporter:Dipl.-Chem. Melanie Mechler ;Dr. René Peters
Angewandte Chemie International Edition 2015 Volume 54( Issue 35) pp:10303-10307
Publication Date(Web):
DOI:10.1002/anie.201502930
Abstract
Diastereodivergency is a challenge for catalytic asymmetric synthesis. For many reaction types, the generation of one diastereomer is inherently preferred, while the other diastereomers are not directly accessible with high efficiency and require circuitous synthetic approaches. Overwriting the inherent preference by means of a catalyst requires control over the spatial positions of both reaction partners. We report a novel polyfunctional catalyst type in which a NiII-bis(phenoxyimine) unit, free hydroxy groups, and an axially chiral bisimidazolium entity participate in the stereocontrol of the direct 1,4-addition of oxindoles to nitroolefins. Both epimers of the 1,4-adduct are accessible in excess on demand by changes to the ligand constitution and configuration. As the products have been reported to be valuable precursors to indole alkaloids, this method should allow access to their epimeric derivatives.
Co-reporter:Dipl.-Chem. Tina Hellmuth;Dr. Wolfgang Frey ;Dr. René Peters
Angewandte Chemie 2015 Volume 127( Issue 9) pp:2829-2833
Publication Date(Web):
DOI:10.1002/ange.201410933
Abstract
Isoxazolinone bilden eine Klasse von Heterocyclen, die häufig bei der Entwicklung von potentiellen neuen Wirkstoffen Verwendung findet. Die cyclische Oximester-Einheit ist außerdem synthetisch wertvoll, da sie funktionelle “Stellschrauben” beinhaltet, die den Zugang zu einer Vielzahl von wertvollen Verbindungsklassen ermöglichen, welche durch andere Methoden nur schwer zugänglich sind. Dennoch sind asymmetrische Methoden zu chiralen Isoxazolinonen offenkundig selten. Hiermit stellen wir die erste katalytische, asymmetrische Alkylierung von Isoxazolinonen zur Bildung von all-C-substituierten quartären Stereozentren vor. Die gegenwärtigen Studien wurden durch die Frage nach der Kontrolle der Regioselektivität aufgrund der Konkurrenz von verschiedenen nukleophilen Positionen angetrieben. Die Untersuchung einer direkten 1,4-Addition ließ erkennen, dass ein sterisch anspruchsvoller palladacyclischer Katalysator die Reaktivität in Abwesenheit einer Base nahezu ausschließlich zum nukleophilen C-Atom steuert, während gleichzeitig hohe Enantioselektivitäten und TONs von bis zu 1900 erreicht werden.
Co-reporter:Tina Hellmuth, Stefan Rieckhoff, Marcel Weiss, Konstantin Dorst, Wolfgang Frey, and René Peters
ACS Catalysis 2014 Volume 4(Issue 6) pp:1850
Publication Date(Web):May 7, 2014
DOI:10.1021/cs500393x
Cooperative asymmetric catalysts often offer advantages in terms of activity, stereoselectivity, and generality as compared to more traditional single point activation catalysts. In cooperative bimetallic catalysis, the intermetallic distance is a crucial parameter for the outcome of a reaction and an optimal synergy of both metal centers. We have recently developed a number of catalytic asymmetric reactions, which are efficiently catalyzed by a planar chiral ferrocene based bispalladacycle and for which the cooperativity of two Pd centers has already been demonstrated. To get more insight into the role of the Pd/Pd distance in such metallocene bismetallacycles, in the present study a corresponding ruthenocene based Pd2-complex has been prepared by the first direct diastereoselective biscyclopalladation of a chiral ruthenocene ligand. In addition, the first highly diastereoselective direct monocyclopalladation of a homochiral ruthenocene is reported. The effect of the increased Cp/Cp distance within the ruthenocene bispalladacycle has been examined in four catalytic asymmetric applications: the aza-Claisen rearrangement of Z-configured allylic N-aryltrifluoroacetimidates, the direct 1,4-addition of α-cyanoacetates to enones, a tandem azlactone formation/1,4-addition to enones and a tandem reaction to form quaternary α-aminosuccinimides by in situ azlactone formation, 1,4-addition to a nitroolefin, and a Nef-type nitro-to-carbonyl transformation as key steps. For each reaction studied, it was found that with some substrates the ferrocene based catalyst is superior, whereas for other substrates the ruthenocene backbone is more favorable. The ruthenocene based bispalladacycle can thus be considered to be a useful and complementary alternative for cooperative bimetallic catalysis.Keywords: 1,4-addition; cyclopalladation; imidazoline; rearrangement; ruthenocene
Co-reporter:Mavila Sudheendran, Simon H. Eitel, Stefan Naumann, Michael R. Buchmeiser and René Peters
New Journal of Chemistry 2014 vol. 38(Issue 11) pp:5597-5607
Publication Date(Web):24 Sep 2014
DOI:10.1039/C4NJ01253D
The anchoring of well-defined molecular catalysts on a surface is an attractive strategy to develop sustainable catalytic processes. This article describes the first syntheses of monolith-supported ferrocene palladacycle catalysts. Monolithic supports were prepared via ring-opening metathesis polymerization (ROMP) using the “1st generation Grubbs catalyst”. Fluorinated carboxylates were surface-grafted utilizing living Ru-termini. The immobilization of the palladacycles onto the monolithic support was accomplished by ligand substitution on the fluorinated carboxylates of the graft polymer. An investigation of these supported catalysts on the efficiency and reusability under different reaction conditions in a direct Michael addition generating a quaternary C-atom is reported. Whereas stereoselectivity was found to be significantly lower than in a comparable homogeneous system, Pd-leaching was not detected in all analyzed samples indicating a permanently immobilized catalyst system.
Co-reporter:Melanie Mechler, Wolfgang Frey, and René Peters
Organometallics 2014 Volume 33(Issue 19) pp:5492-5508
Publication Date(Web):September 4, 2014
DOI:10.1021/om500762r
Salen and NHC ligands are among the most important ligands for homogeneous catalysis. We have recently reported bimetallic complexes, in which both motifs have been merged for the first time. However, the intermetallic distances, which play a crucial role for cooperative bimetallic catalysis, were probably not appropriate in these first-generation hybrid catalysts. To generate heterobimetallic salen/NHC hybrid complexes with intermetallic distances suitable for cooperative catalysis, chiral macrocyclic hybrid ligands featuring a salen and two linked NHC donor moieties have been prepared in the present study. For the ligand formation, chiral enantiopure diamines as well as chiral enantiopure bisimidazoles were employed, and a matched/mismatched situation was found depending on the configuration of both chirality sources. Regioselective complexation of Zn(II), Ni(II), and Pd(II) by the salen N2O2 coordination sphere was efficiently accomplished. Subsequent coordination of the NHC units was achieved for Ag(I), Cu(I), Au(I), and Pd(II), in the latter case by oxidative transmetalation with Pd2(dba)3. X-ray crystal structure analyses for Ni/Ag2 and Pd/Ag2 complexes show strongly puckered macrocycles, in which one of the NHC-bound Ag(I) centers is in close proximity to the salen-bound Ni(II) or Pd(II) centers and in which this Ag(I) apparently interacts with both salen O-donor atoms. Preliminary data for the 1,4-addition of an oxindole to a nitroolefin and for the Conia-ene reaction of an α-cyanoacetate are reported.
Co-reporter:Dipl.-Chem. Johannes Moritz Bauer;Dr. Wolfgang Frey ;Dr. René Peters
Angewandte Chemie International Edition 2014 Volume 53( Issue 29) pp:7634-7638
Publication Date(Web):
DOI:10.1002/anie.201403090
Abstract
A regio- and enantioselective tandem reaction is reported capable of directly transforming readily accessible achiral allylic alcohols into chiral sulfonyl-protected allylic amines. The reaction is catalyzed by the cooperative action of a chiral ferrocene palladacycle and a tertiary amine base and combines high step-economy with operational simplicity (e.g. no need for inert-gas atmosphere or catalyst activation). Mechanistic studies support a PdII-catalyzed [3,3] rearrangement of allylic carbamates—generated in situ from the allylic alcohol and an isocyanate—as the key step, which is followed by a decarboxylation.
Co-reporter:Dipl.-Chem. Johannes Moritz Bauer;Dr. Wolfgang Frey ;Dr. René Peters
Angewandte Chemie 2014 Volume 126( Issue 29) pp:7764-7768
Publication Date(Web):
DOI:10.1002/ange.201403090
Abstract
Eine regio- und enantioselektive Tandemreaktion, die leicht zugängliche achirale Allylalkohole direkt in chirale sulfonylgeschützte Allylamine überführt, wird beschrieben. Die Reaktion wird durch Kooperation eines chiralen Ferrocen-Palladacyclus und einer tertiären Amin-Base katalysiert und kombiniert hohe Stufenökonomie mit leichter Handhabung (ohne Inertgasatmosphäre oder Katalysatoraktivierung). Mechanistische Studien stützen eine PdII-katalysierte [3,3]-Umlagerung eines Allylcarbamats – in situ erzeugt aus einem Allylalkohol und einem Isocyanat – als Schlüsselschritt, dem eine Decarboxylierung folgt.
Co-reporter:Marta Ayerbe Garcia, Wolfgang Frey, and René Peters
Organometallics 2014 Volume 33(Issue 4) pp:1068-1078
Publication Date(Web):February 6, 2014
DOI:10.1021/om500049f
Functionalized 1′,2′,3′,4′,5′-pentaphenylferrocenes are known as valuable ligands and catalysts. Planar chiral derivatives—e.g. Fu’s pentaphenylferrocene fused pyridines or palladacycles developed in our group—have previously been described to be very efficient asymmetric catalysts for a number of applications. Nevertheless, protocols for diastereoselective ortho lithiations of 1′,2′,3′,4′,5′-pentaphenylferrocenes are still unknown. Such protocols could significantly increase the arsenal of accessible planar chiral pentaphenylferrocenes. In this full paper, we describe such diastereoselective ortho lithiations of pentaphenylferrocenyloxazolines. Both possible diastereomers can be produced in almost diastereomerically pure form depending on the choice of a Lewis base additive, which is required in the lithiation step for a sufficient reactivity. This development was utilized to prepare a set of planar chiral pentaphenylferrocene-based phosphino-oxazoline ligands. The latter were investigated in Pd-catalyzed allylic substitution reactions with cyclic allylic acetates to allow for a first comparison with related planar chiral ligands and provided good enantioselectivity with this challenging substrate class.
Co-reporter:Simon H. Eitel, Sascha Jautze, Wolfgang Frey and René Peters
Chemical Science 2013 vol. 4(Issue 5) pp:2218-2233
Publication Date(Web):22 Mar 2013
DOI:10.1039/C3SC50419K
The direct asymmetric conjugate addition of α-cyanoacetates to enones generating densely functionalized α-amino acid precursors with adjacent quaternary and tertiary stereocenters is described comparing mono- and bis-palladacycle catalysts. This edge article features the complementary value of mono- and bimetallic catalysis in a case study using related catalyst systems. Different major diastereomers of the 1,4-addition products are formed by the use of the planar chiral mono- and bimetallic catalyst systems and provide access to epimeric amino acid derivatives. Both catalyst types require the use of a Brønsted acid (HOAc) as a co-catalyst to avoid an undesired β-hydride elimination. Kinetic studies show that the C–C bond forming step takes place almost instantaneously with the bis-palladium complex after productive substrate coordination. This extraordinarily high reactivity for an elementary step generating a sterically demanding linkage of a quaternary and a tertiary stereocenter stresses the cooperativity of both metal centers.
Co-reporter:Dipl.-Chem. Manuel Weber;Dr. Wolfgang Frey ;Dr. René Peters
Angewandte Chemie International Edition 2013 Volume 52( Issue 50) pp:13223-13227
Publication Date(Web):
DOI:10.1002/anie.201307334
Co-reporter:Melanie Mechler, Katja Latendorf, Wolfgang Frey, and René Peters
Organometallics 2013 Volume 32(Issue 1) pp:112-130
Publication Date(Web):December 18, 2012
DOI:10.1021/om3008629
salen ligands and N-heterocyclic carbenes (NHC) are among the most abundant structural ligand motifs for metal catalysis. In this article we present a modular approach to chiral ligands merging both ligand motifs for the preparation of bimetallic catalysts, in which one metal (M1) is coordinated by the salen moiety and the other metal (M2) binds to two NHCs. After selective complexation of M1 = PdII, NiII ion into the salen N2O2 coordination site, heterobimetallic M1/Ag(I) complexes were synthesized which could be further utilized for the preparation of homo- and heterobimetallic M1/PdII complexes by an oxidative transmetalation to Pd(0) as a key step of the catalyst synthesis. The structures of the bimetallic complexes and the intermetallic distances strongly depend on the counterions of M2 = PdII, as revealed by X-ray and UV–vis studies. In the absence of an anionic ligand with suitable Lewis basicity the salen oxygen atoms serve as bridging ligands for both metals. A preliminary investigation into catalysis showed that the complexes are capable of catalyzing the 1,4-addition of oxindoles to 2-nitrostyrene.
Co-reporter:Manuel Weber, Johannes E. M. N. Klein, Burkhard Miehlich, Wolfgang Frey, and René Peters
Organometallics 2013 Volume 32(Issue 20) pp:5810-5817
Publication Date(Web):May 28, 2013
DOI:10.1021/om400360d
Monomeric ferrocene bis-imidazoline bis-palladacycles (FBIP) have recently been reported to be efficient bimetallic catalysts in different sorts of asymmetric reactions by the cooperation of two Pd(II) centers. A crucial parameter regarding the efficiency of reactions catalyzed in a bimetallic mode is—in general—the intermetallic distance of both catalytically relevant metal centers. In this article we describe the structural elucidation of the monomeric FBIP catalyst type (usually generated in situ from a catalytically inactive dimeric chloride bridged precatalyst) by X-ray crystal structure analysis. Two dicationic monomeric complexes are compared to a neutral complex. The solid-state structures reveal varying Pd–Pd distances ranging from 3.15 to 5.27 Å for the doubly charged complexes, whereas for the neutral complex a distance of 3.28 Å has been found. This variability is supposed to be one of the key advantages of a ferrocene backbone in a bimetallic catalyst system, since the Fe–Cp bonds allow the bimetallic complex to readily open and close like a pair of scissors, employing just a few degrees of rotational freedom. In addition, on the basis of the nature of the reported catalyst species, we suggest that a permanent switch among neutral and mono- and dicationic catalyst species by a Brønsted acid such as acetic acid might facilitate different elementary steps in a catalytic cycle. By DFT calculations we have found that weak d8–d8 interactions contribute to short Pd–Pd distances but are less important than dispersive interactions, which can even overcome the Coulombic repulsion of two cationic palladium centers.
Co-reporter:Dipl.-Chem. Manuel Weber;Dr. Wolfgang Frey ;Dr. René Peters
Angewandte Chemie 2013 Volume 125( Issue 50) pp:13465-13469
Publication Date(Web):
DOI:10.1002/ange.201307334
Co-reporter:Manuel Weber;Dr. Wolfgang Frey ;Dr. René Peters
Chemistry - A European Journal 2013 Volume 19( Issue 25) pp:8342-8351
Publication Date(Web):
DOI:10.1002/chem.201300224
Abstract
Spirocyclic azlactones are shown to be useful precursors of cyclic quaternary amino acids, such as the constrained cyclohexane analogues of phenylalanine. These compounds are of interest as building blocks for the synthesis of artificial peptide analogues with controlled folds in the peptide backbone. They were prepared in the present study by a step- and atom-economic catalytic asymmetric tandem approach, requiring two steps starting from N-benzoyl glycine and divinylketones. The key of this protocol is the enantioselective formation of the azlactone spirocycles, which involves a PdII-catalyzed double 1,4-addition of an in situ generated azlactone intermediate to the dienone (a formal [5+1] cycloaddition). As the catalyst, a planar chiral ferrocene bispalladacycle was used. Mechanistic studies suggest a monometallic reaction pathway. Although the diastereoselectivity was found to be moderate, the enantioselectivity is usually high for the formation of the azlactone spirocycles, which contain up to three contiguous stereocenters. Spectroscopic studies have shown that the spirocycles often prefer a twist over a chair conformation of the cyclohexanone moiety.
Co-reporter:Manuel Weber;Wolfgang Frey ;René Peters
Advanced Synthesis & Catalysis 2012 Volume 354( Issue 8) pp:1443-1449
Publication Date(Web):
DOI:10.1002/adsc.201200085
Abstract
Masked and activated highly enantioenriched α,α-disubstituted α-amino acids with an additional adjacent stereocenter were formed by a tandem reaction involving five steps using racemic unprotected amino acid substrates. Key step is the 1,4-addition of in-situ generated azlactones to a broad number of enones. The products of this step-economic route can, e.g., be useful for a divergent and rapid access to biologically interesting unnatural glutamic acid derivatives.
Co-reporter:Manuel Weber and René Peters
The Journal of Organic Chemistry 2012 Volume 77(Issue 23) pp:10846-10855
Publication Date(Web):November 29, 2012
DOI:10.1021/jo302177t
A multicomponent reaction is reported generating highly enantioenriched and diastereomerically pure quaternary amino acid derivatives via 1,4-addition of azlactones to enones. The azlactone intermediates are generated in situ from unprotected α-amino acids and acetic anhydride. Previous attempts using bis-palladacycle catalysts required the use of a large excess of benzoic anhydride (which is very difficult to remove from the products), since acetic anhydride provided regioisomeric product mixtures. Key for the high regioselectivity is a pentaphenylferrocene monopalladacycle catalyst.
Co-reporter:Marcel Weiss, Wolfgang Frey, and René Peters
Organometallics 2012 Volume 31(Issue 17) pp:6365-6372
Publication Date(Web):August 22, 2012
DOI:10.1021/om300600v
Ligand exchange reactions are usually slower for Pt(II) in comparison to Pd(II) centers. Mainly for that reason Pt(II) catalysts have often shown a reduced catalytic activity as compared to their Pd(II) counterparts. We are interested in the question if this inherently slower ligand exchange might also provide a chance for heterobimetallic catalysts to accomplish an improved catalytic performance with substrates in which the reactive center has a lower binding constant than an additional Lewis basic moiety. For that purpose we have prepared the first diastereo- and enantiomerically pure mixed pallada-/platinacycles based on ferrocene. These complexes have been prepared by sequential direct diastereoselective cycloplatination and cyclopalladation. The investigation of the asymmetric aza-Claisen rearrangement of Z-configured trifluoroacetimidates showed that a heterodinuclear Pt–Pd bis-metallacycle is an excellent catalyst for this reaction type, in general allowing for very high enantioselectivities. Moreover, at a slightly elevated temperature (55 °C), the heterodinuclear platina-/palladacycle could in certain cases outperform the corresponding bis-Pd complex, previously known to be the by far most active highly enantioselective catalyst for the rearrangement of Z-configured trifluoroacetimidates. This effect, which might be surprising at first sight due to the low efficiency of other Pt catalysts for aza-Claisen rearrangements, might be explained by an enhanced lifetime of a productive monodentate olefin coordination of the substrate at the Pt center due to slower ligand exchange processes.
Co-reporter:Dipl.-Chem. Manuel Weber;Dr. Sascha Jautze;Dr. Wolfgang Frey ;Dr. René Peters
Chemistry - A European Journal 2012 Volume 18( Issue 46) pp:14792-14804
Publication Date(Web):
DOI:10.1002/chem.201202455
Abstract
The development and further evolution of the first catalytic asymmetric conjugate additions of azlactones as activated amino acid derivatives to enones is described. Whereas the first-generation approach started from isolated azlactones, in the second-generation approach the azlactones could be generated in situ starting from racemic N-benzoylated amino acids. The third evolution stage could make use of racemic unprotected α-amino acids to directly form highly enantioenriched and diastereomerically pure masked quaternary amino acid products bearing an additional tertiary stereocenter. The step-economic transformations were accomplished by cooperative activation by using a robust planar chiral bis-Pd catalyst, a Brønsted acid (HOAc or BzOH; Ac=acetyl, Bz=benzoyl), and a Brønsted base (NaOAc). In particular the second- and third-generation approaches provide a rapid and divergent access to biologically interesting unnatural quaternary amino acid derivatives from inexpensive bulk chemicals. In that way highly enantioenriched acyclic α-amino acids, α-alkyl proline, and α-alkyl pyroglutamic acid derivatives could be prepared in diastereomerically pure form. In addition, a unique way is presented to prepare diastereomerically pure bicyclic dipeptides in just two steps from unprotected tertiary α-amino acids.
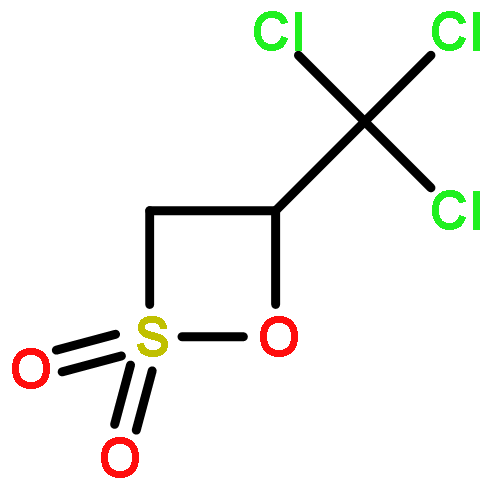
Co-reporter:Dr. Florian M. Koch;Dr. René Peters
Chemistry - A European Journal 2011 Volume 17( Issue 13) pp:3679-3692
Publication Date(Web):
DOI:10.1002/chem.201003542
Abstract
The first catalytic asymmetric synthesis of β-sultones is reported. This development has enabled a rapid access to a number of highly enantioenriched biologically interesting sulfonyl and sulfinyl compound classes, which makes use of the inherent ring strain of the four-membered heterocycles. The products possess either two vicinal stereocenters, such as in β-hydroxy-sulfonamides, -sulfonates, -sulfones, -sulfonic acids, -sulfinic acids, γ-sultines, and γ-sultones or a single stereocenter, such as in α-branched alkyl or allyl sulfonic acids. This work also represents the first application of sulfene intermediates in asymmetric catalysis. The reactivity of a sulfene normally acting as an electrophile could be reverted by the formation of a nucleophilic zwitterionic sulfene–amine adduct. To achieve a combination of high enantioselectivity and reactivity, cooperative catalytic action of a chiral nucleophilic tertiary amine (the cinchona alkaloid derivative diydroquinine 2,5-diphenyl-4,6-pyrimidinediyl diether ((DHQ)2PYR)) and Bi(OTf)3 or In(OTf)3 was of primary importance.
Co-reporter:Manuel Weber ; Sascha Jautze ; Wolfgang Frey ;René Peters
Journal of the American Chemical Society 2010 Volume 132(Issue 35) pp:12222-12225
Publication Date(Web):August 17, 2010
DOI:10.1021/ja106088v
Cooperative activation by a soft bimetallic catalyst, a hard Brønsted acid, and a hard Brønsted base has allowed the formation of highly enantioenriched, diastereomerically pure masked α-amino acids with adjacent quaternary and tertiary stereocenters in a single reaction starting from racemic N-benzoylated amino acids. The products can, for example, be used to prepare bicyclic dipeptides.
Co-reporter:PaoloS. Tiseni Dr.;René Peters Dr.
Chemistry - A European Journal 2010 Volume 16( Issue 8) pp:2503-2517
Publication Date(Web):
DOI:10.1002/chem.200902896
Abstract
Previously unexplored enantiopure zwitterionic ammonium dienolates have been utilized in this work as reactive intermediates that act as diene components in hetero-Diels–Alder reactions (HDAs) with aldehydes to produce optically active δ-lactones, subunits of numerous bioactive products. The dienolates were generated in situ from E/Z mixtures of α,β-unsaturated acid chlorides by use of a nucleophilic quinidine derivative and Sn(OTf)2 as co-catalyst. The latter component was not directly involved in the cycloaddition step with aldehydes and simply facilitated the formation of the reactive dienolate species. The scope of the cycloaddition was considerably improved by use of a complex formed from Er(OTf)3 and a simple commercially available norephedrine-derived ligand that tolerated a broad range of aromatic and heteroaromatic aldehydes for a cooperative bifunctional Lewis-acid-/Lewis-base-catalyzed reaction, providing α,β-unsaturated δ-lactones with excellent enantioselectivities. Mechanistic studies confirmed the formation of the dienolate intermediates for both catalytic systems. The active ErIII complex is most likely a monomeric species. Interestingly, all lanthanides can catalyze the title reaction, but the efficiency in terms of yield and enantioselectivity depends directly on the radius of the LnIII ion. Similarly, use of the pseudolanthanides ScIII and YIII also resulted in product formation, whereas the larger LaIII and other transition metal salts, as well as main group metal salts, proved to be inefficient. In addition, various synthetic transformations of 6-CCl3- or 4-silyl-substituted α,β-unsaturated δ-lactones, giving access to a number of valuable δ-lactone building blocks, were investigated.
Co-reporter:Thomas Kull Dr.;José Cabrera Dr.;René Peters Dr.
Chemistry - A European Journal 2010 Volume 16( Issue 30) pp:9132-9139
Publication Date(Web):
DOI:10.1002/chem.201000840
Abstract
The development of the first trans-selective catalytic asymmetric [2+2] cyclocondensation of acyl halides with aliphatic aldehydes furnishing 3,4-disubstituted β-lactones is described. This work made use of a new strategy within the context of asymmetric dual activation catalysis: it combines the concepts of Lewis acid and organic aprotic ion pair catalysis in a single catalyst system. The methodology could also be applied to aromatic aldehydes and offers broad applicability (29 examples). The utility was further demonstrated by nucleophilic ring-opening reactions that provide highly enantiomerically enriched anti-aldol products.
Co-reporter:René Peters Dr.;Zhuo-qun Xin Dr. ;Frank Maier
Chemistry – An Asian Journal 2010 Volume 5( Issue 8) pp:1770-1774
Publication Date(Web):
DOI:10.1002/asia.201000386
Co-reporter:Sascha Jautze, Stefan Diethelm, Wolfgang Frey and René Peters
Organometallics 2009 Volume 28(Issue 7) pp:2001-2004
Publication Date(Web):March 12, 2009
DOI:10.1021/om900137c
An axially chiral reactive precursor—a trans-configured Pd(II) chelate complex—toward the planar chiral bis-palladacycle FBIP-Cl, which was recently shown to be an excellent enantioselective catalyst for both aza-Claisen rearrangements and direct Michael additions, has been identified and allows us to explain the stereochemical outcome of the first reported direct diastereoselective bis-cyclopalladation. In contrast to the trans-configured chelate complex, its cis-configured counterpart turned out to be a dead end under the initial cyclopalladation conditions and required harsher conditions for C−H activation.
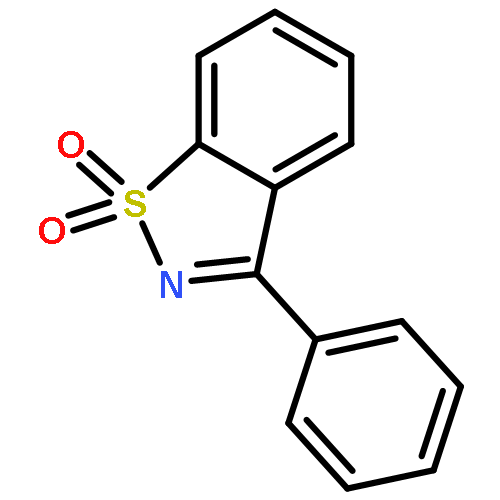
Co-reporter:Haoxi Huang Dr.;René Peters Dr.
Angewandte Chemie International Edition 2009 Volume 48( Issue 3) pp:604-606
Publication Date(Web):
DOI:10.1002/anie.200804944
Co-reporter:Marian Zajac Dr.;René Peters Dr.
Chemistry - A European Journal 2009 Volume 15( Issue 33) pp:8204-8222
Publication Date(Web):
DOI:10.1002/chem.200900496
Abstract
β-Sultams, biologically interesting sulfonyl analogues of β-lactams, have been prepared by an organocatalytic asymmetric formal [2+2]-cycloaddition approach of non-nucleophilic imines with alkyl sulfonyl chlorides. In the case of very electron poor N-tosyl imines derived from chloral or ethylglyoxylate, this reaction type was catalyzed by cinchona alkaloids providing the heterocycles in high yield, with good diastereoselectivity and up to 94 % ee. Mechanistic investigations suggested that the product formation proceeded via a zwitterionic imine–catalyst adduct. The scope was significantly extended by 2-pyridylsulfonyl imines derived from non-activated aromatic aldehydes employing Yb(OTf)3 as Lewis acid cocatalyst. The synthetic value of the strained enantioenriched β-sultams was demonstrated by smooth nucleophilic ring opening reactions with O-, N- and C-nucleophiles yielding a variety of acyclic β-aminosulfonyl (taurine) derivatives (sulfonates, sulfonamides, sulfones) without racemization or epimerization.
Co-reporter:DanielF. Fischer Dr.;Assem Barakat;Zhuo-qun Xin;MatthiasE. Weiss;René Peters Dr.
Chemistry - A European Journal 2009 Volume 15( Issue 35) pp:8722-8741
Publication Date(Web):
DOI:10.1002/chem.200900712
Abstract
Systematic studies have been performed to develop highly efficient catalysts for the asymmetric aza-Claisen rearrangement of trihaloacetimidates. Herein, we describe the stepwise development of these catalyst systems involving four different catalyst generations finally resulting in the development of a planar chiral pentaphenylferrocenyl oxazoline palladacycle. This complex is more reactive and has a broader substrate tolerance than all previously known catalyst systems for asymmetric aza-Claisen rearrangements. Our investigations also reveal that subtle changes can have a big impact on the activity. With the enhanced catalyst activity, the asymmetric aza-Claisen rearrangement has a very broad scope: the methodology not only allows the formation of highly enantioenriched primary allylic amines, but also secondary and tertiary amines; allylic amines with N-substituted quaternary stereocenters are conveniently accessible as well. The reaction conditions tolerate many important functional groups, thus providing stereoselective access to valuable functionalized building blocks, for example, for the synthesis of unnatural amino acids. Our results suggest that face-selective olefin coordination is the enantioselectivity-determining step, which is almost exclusively controlled by the element of planar chirality.
Co-reporter:Haoxi Huang Dr.;René Peters Dr.
Angewandte Chemie 2009 Volume 121( Issue 3) pp:612-615
Publication Date(Web):
DOI:10.1002/ange.200804944
Co-reporter:Sascha Jautze;René Peters Dr.
Angewandte Chemie 2008 Volume 120( Issue 48) pp:9424-9429
Publication Date(Web):
DOI:10.1002/ange.200803539
Co-reporter:Sascha Jautze;René Peters Dr.
Angewandte Chemie International Edition 2008 Volume 47( Issue 48) pp:9284-9288
Publication Date(Web):
DOI:10.1002/anie.200803539
Co-reporter:Simon H. Eitel, Sascha Jautze, Wolfgang Frey and René Peters
Chemical Science (2010-Present) 2013 - vol. 4(Issue 5) pp:NaN2233-2233
Publication Date(Web):2013/03/22
DOI:10.1039/C3SC50419K
The direct asymmetric conjugate addition of α-cyanoacetates to enones generating densely functionalized α-amino acid precursors with adjacent quaternary and tertiary stereocenters is described comparing mono- and bis-palladacycle catalysts. This edge article features the complementary value of mono- and bimetallic catalysis in a case study using related catalyst systems. Different major diastereomers of the 1,4-addition products are formed by the use of the planar chiral mono- and bimetallic catalyst systems and provide access to epimeric amino acid derivatives. Both catalyst types require the use of a Brønsted acid (HOAc) as a co-catalyst to avoid an undesired β-hydride elimination. Kinetic studies show that the C–C bond forming step takes place almost instantaneously with the bis-palladium complex after productive substrate coordination. This extraordinarily high reactivity for an elementary step generating a sterically demanding linkage of a quaternary and a tertiary stereocenter stresses the cooperativity of both metal centers.
Co-reporter:Marta Ayerbe Garcia, Wolfgang Frey, Mark R. Ringenberg, Max Schwilk and René Peters
Chemical Communications 2015 - vol. 51(Issue 94) pp:NaN16809-16809
Publication Date(Web):2015/10/05
DOI:10.1039/C5CC07018J
Oxidation of Au(I) in the presence of Fe(II) allowed for the synthesis of unique dinuclear ferrocenyl Au(II) complexes via the first reported enantiopure planar chiral ferrocenyl Au(I) complex. (Spectro)electrochemical studies show that oxidation at Fe(II) is favoured, but DFT studies suggest that the energy differences for oxidation of one or the other metal should be quite small.
Co-reporter:J. Moritz Bauer and René Peters
Catalysis Science & Technology (2011-Present) 2015 - vol. 5(Issue 4) pp:NaN2346-2346
Publication Date(Web):2015/02/06
DOI:10.1039/C4CY01749H
A streamlined synthetic access to enantiomerically pure allylic amines as advanced and valuable synthetic building blocks is important for pharmaceutical sciences. The rearrangement of allylic trihaloacetimidates is known as an attractive method to furnish branched chiral allylic trihaloacetamides with high levels of enantio- and regioselectivity. In the present article we report our studies on the catalytic asymmetric rearrangement of the corresponding non-halogenated acetimidates, which might provide economic advantages by avoiding CX3 groups. The regioselective title reaction proceeds with high levels of enantioselectivity, provides high yields and requires only low catalyst loadings. In addition, the generated N-acetyl and N-phenacetyl functionalities offer the option of a subsequent mild enzymatic amide hydrolysis to get access to nearly enantiopure allylic amines.
Co-reporter:Florian Broghammer, Daniel Brodbeck, Thorsten Junge and René Peters
Chemical Communications 2017 - vol. 53(Issue 6) pp:NaN1159-1159
Publication Date(Web):2016/12/23
DOI:10.1039/C6CC09774J
Epoxide desymmetrizations by bromide are very rare despite the large synthetic potential of chiral bromohydrins. Herein we present a new concept for epoxide desymmetrizations in which a bifunctional Lewis acid/ammonium salt catalyst allows for efficient enantioselective epoxide ring openings by Br−. With acetylbromide as a Br− source bromohydrin esters are formed.

![N'-[(R)-(4-chlorophenyl)phenylmethyl]-N,N-dimethylsulfamide](http://img.cochemist.com/ccimg/898300/898231-59-9.png)
![N'-[(R)-(4-chlorophenyl)phenylmethyl]-N,N-dimethylsulfamide](http://img.cochemist.com/ccimg/898300/898231-59-9_b.png)
![2-Pyridinesulfonamide, N-[(3-fluorophenyl)methylene]-](http://img.cochemist.com/ccimg/880500/880469-86-3.png)
![2-Pyridinesulfonamide, N-[(3-fluorophenyl)methylene]-](http://img.cochemist.com/ccimg/880500/880469-86-3_b.png)


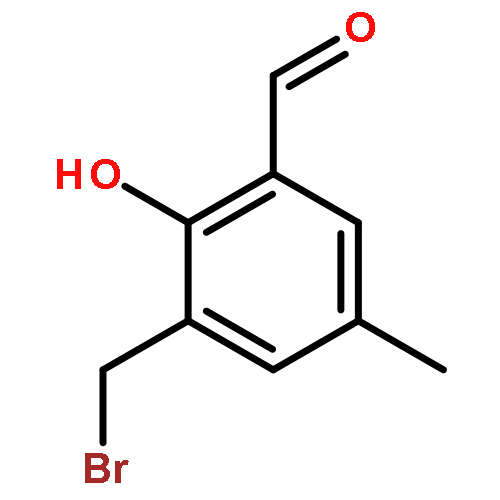
![5(2H)-ISOXAZOLONE, 4-[(4-NITROPHENYL)METHYL]-3-PHENYL-](http://img.cochemist.com/ccimg/863100/863037-44-9.png)
![5(2H)-ISOXAZOLONE, 4-[(4-NITROPHENYL)METHYL]-3-PHENYL-](http://img.cochemist.com/ccimg/863100/863037-44-9_b.png)


![Bicyclo[2.2.1]hept-2-ene, 5-[(3-iodopropoxy)methyl]-, (1R,4R,5R)-rel-](http://img.cochemist.com/ccimg/851800/851777-99-6.png)
![Bicyclo[2.2.1]hept-2-ene, 5-[(3-iodopropoxy)methyl]-, (1R,4R,5R)-rel-](http://img.cochemist.com/ccimg/851800/851777-99-6_b.png)
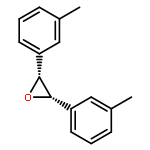
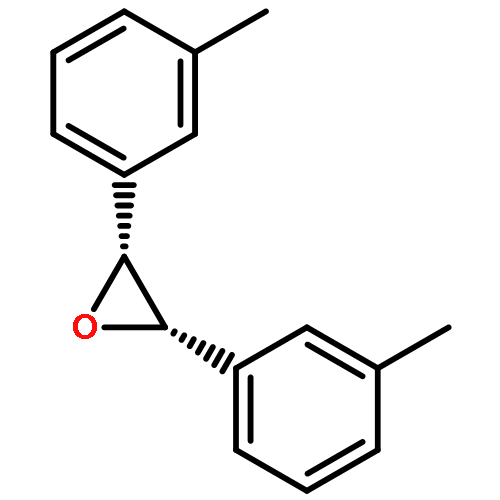
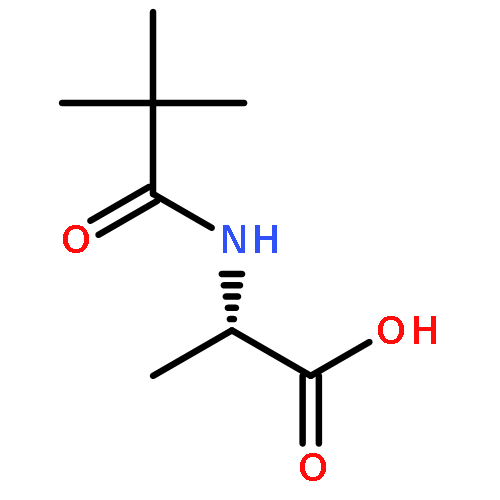
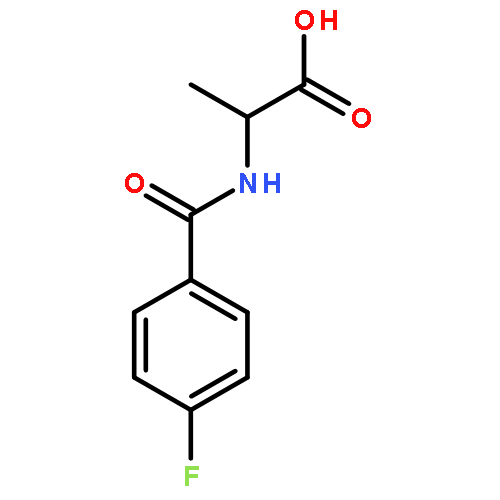
![2-[(4-tert-butylbenzoyl)amino]propanoic acid](http://img.cochemist.com/ccimg/726200/726174-56-7.png)
![2-[(4-tert-butylbenzoyl)amino]propanoic acid](http://img.cochemist.com/ccimg/726200/726174-56-7_b.png)


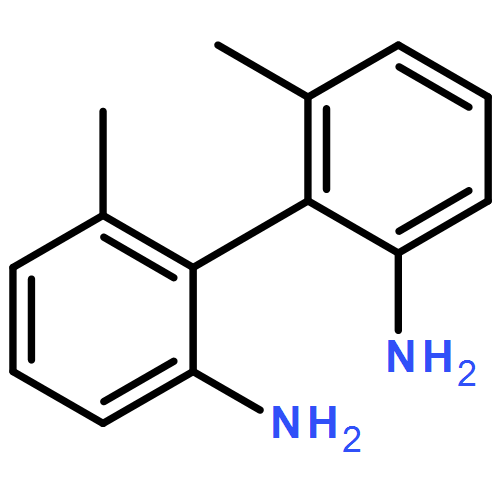
![1H-Imidazole, 1,1'-[(1R,2R)-1,2-diphenyl-1,2-ethanediyl]bis-](http://img.cochemist.com/ccimg/647900/647841-35-8.png)
![1H-Imidazole, 1,1'-[(1R,2R)-1,2-diphenyl-1,2-ethanediyl]bis-](http://img.cochemist.com/ccimg/647900/647841-35-8_b.png)
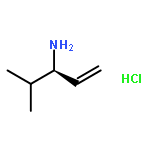
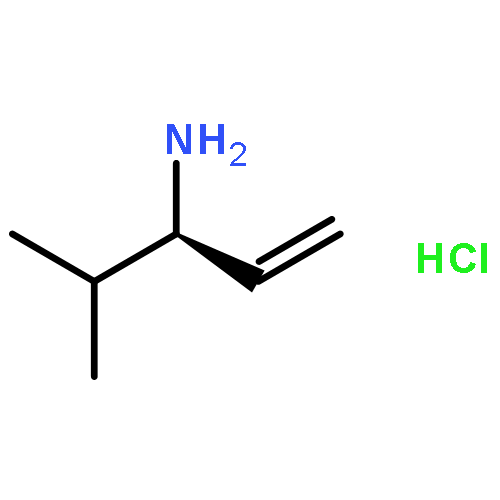
![Benzenamine, N-[(1R)-1-ethenyl-3-methylbutyl]-4-methoxy-](http://img.cochemist.com/ccimg/562900/562870-94-4.png)
![Benzenamine, N-[(1R)-1-ethenyl-3-methylbutyl]-4-methoxy-](http://img.cochemist.com/ccimg/562900/562870-94-4_b.png)
![Benzenamine, 4-methoxy-N-[(1S)-1-methyl-2-propenyl]-](http://img.cochemist.com/ccimg/562900/562870-92-2.png)
![Benzenamine, 4-methoxy-N-[(1S)-1-methyl-2-propenyl]-](http://img.cochemist.com/ccimg/562900/562870-92-2_b.png)
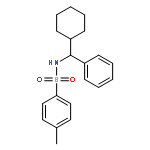
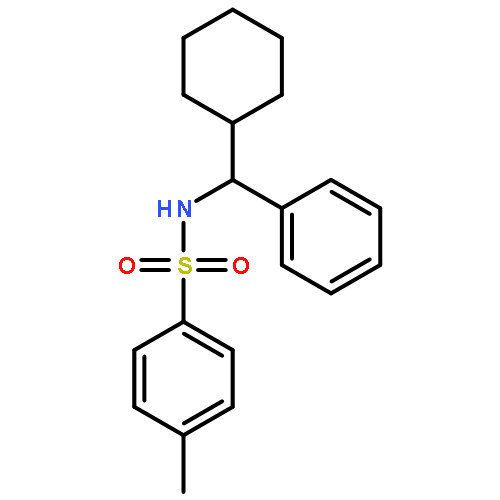

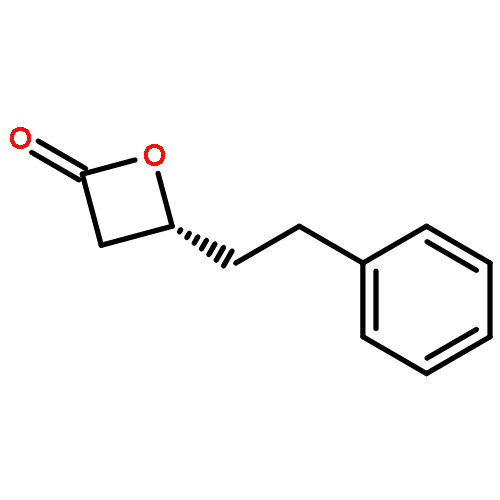
![Benzenesulfonamide, N-[(4-chlorophenyl)phenylmethyl]-4-methyl-](http://img.cochemist.com/ccimg/258300/258277-20-2.png)
![Benzenesulfonamide, N-[(4-chlorophenyl)phenylmethyl]-4-methyl-](http://img.cochemist.com/ccimg/258300/258277-20-2_b.png)
![Benzenesulfonamide, N-[(2-chlorophenyl)phenylmethyl]-4-methyl-](http://img.cochemist.com/ccimg/258300/258277-19-9.png)
![Benzenesulfonamide, N-[(2-chlorophenyl)phenylmethyl]-4-methyl-](http://img.cochemist.com/ccimg/258300/258277-19-9_b.png)
![Benzenesulfonamide, N-[(4-methoxyphenyl)phenylmethyl]-4-methyl-](http://img.cochemist.com/ccimg/258300/258277-16-6.png)
![Benzenesulfonamide, N-[(4-methoxyphenyl)phenylmethyl]-4-methyl-](http://img.cochemist.com/ccimg/258300/258277-16-6_b.png)



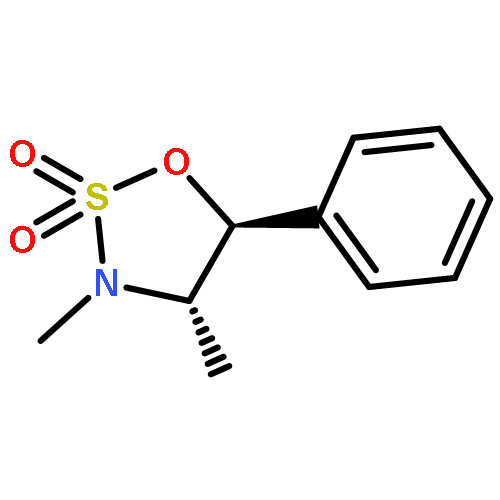
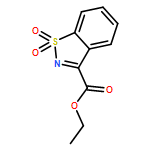
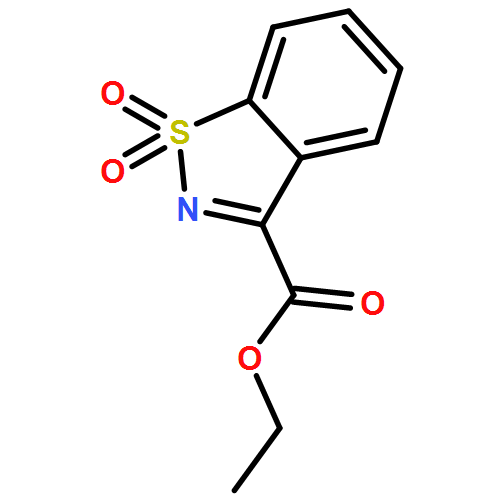
![Cinchonan,9,9''-[(2,5-diphenyl-4,6-pyrimidinediyl)bis(oxy)]bis[10,11-dihydro-6'-methoxy-,(8a,9R)-(8''a,9''R)-](http://img.cochemist.com/ccimg/149900/149820-65-5.png)
![Cinchonan,9,9''-[(2,5-diphenyl-4,6-pyrimidinediyl)bis(oxy)]bis[10,11-dihydro-6'-methoxy-,(8a,9R)-(8''a,9''R)-](http://img.cochemist.com/ccimg/149900/149820-65-5_b.png)
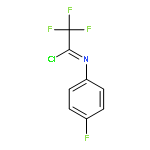
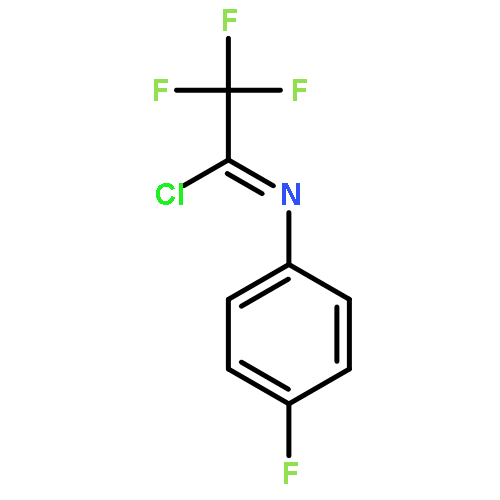
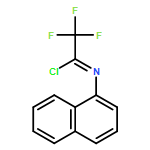
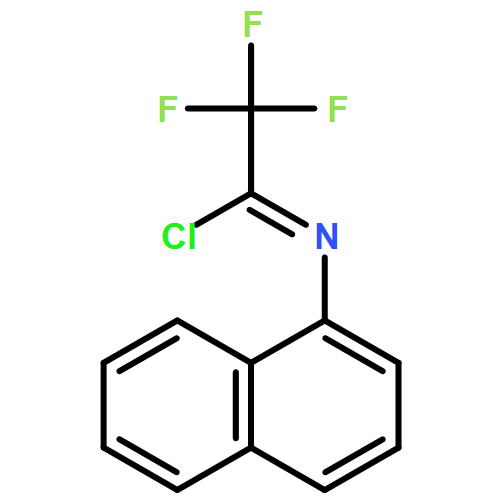
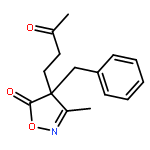


![Cyclohexane, [(1E)-2-nitroethenyl]-](http://img.cochemist.com/ccimg/132900/132839-80-6.png)
![Cyclohexane, [(1E)-2-nitroethenyl]-](http://img.cochemist.com/ccimg/132900/132839-80-6_b.png)
![2-Propen-1-ol, 3-[4-(trifluoromethyl)phenyl]-, (2E)-](/data/chemimg/115900/125617-18-7.png)
![2-Propen-1-ol, 3-[4-(trifluoromethyl)phenyl]-, (2E)-](/data/chemimg/115900/125617-18-7_b.png)
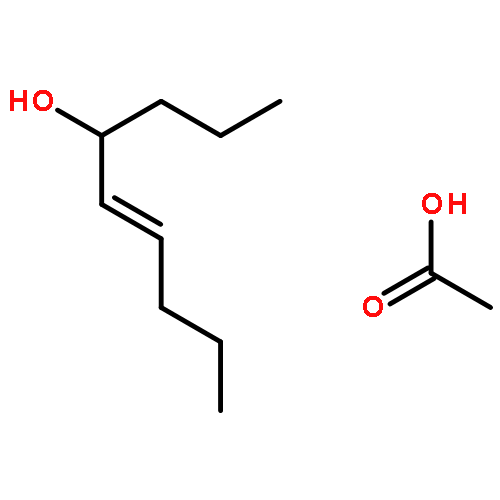

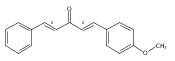
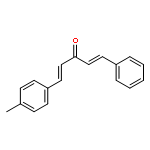
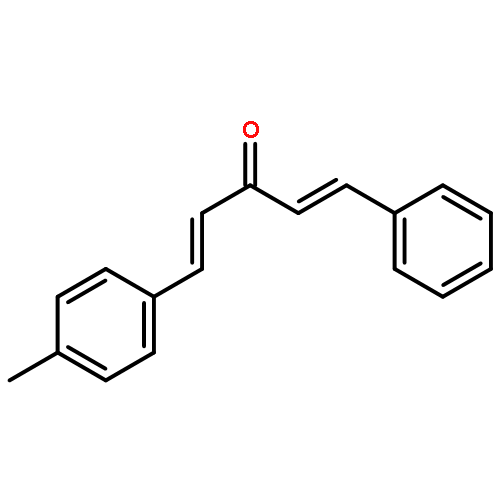
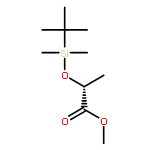
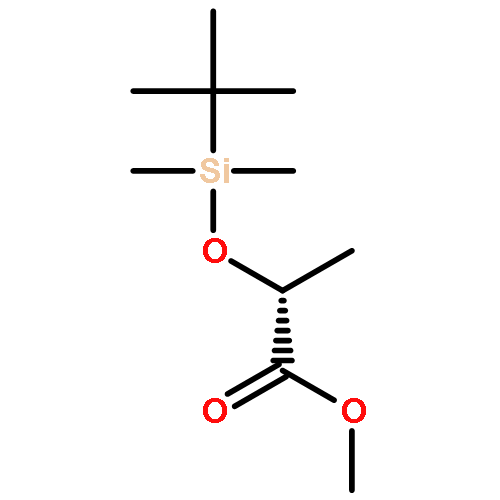
![Propanal, 2-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-, (R)-](http://img.cochemist.com/ccimg/111900/111819-71-7.png)
![Propanal, 2-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-, (R)-](http://img.cochemist.com/ccimg/111900/111819-71-7_b.png)
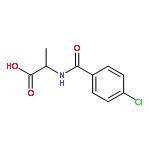
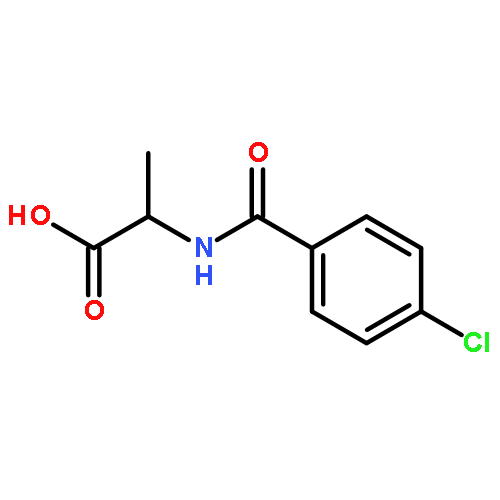
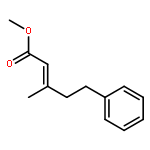
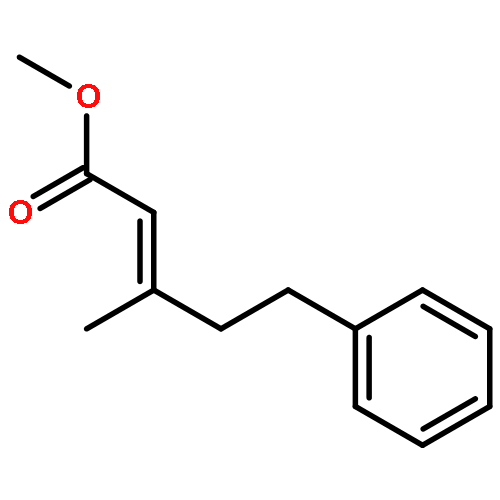
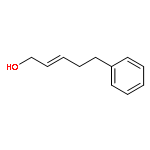
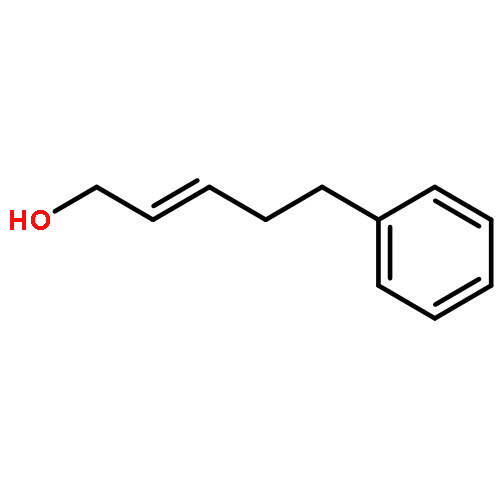

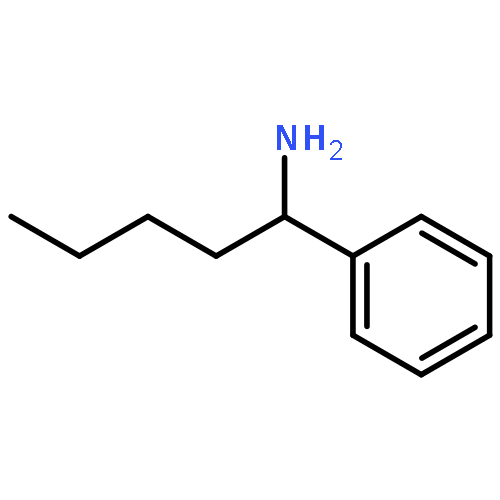
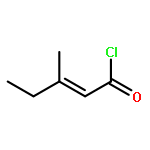
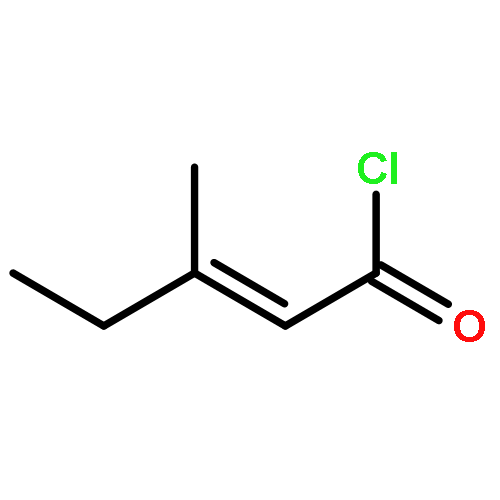
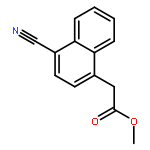
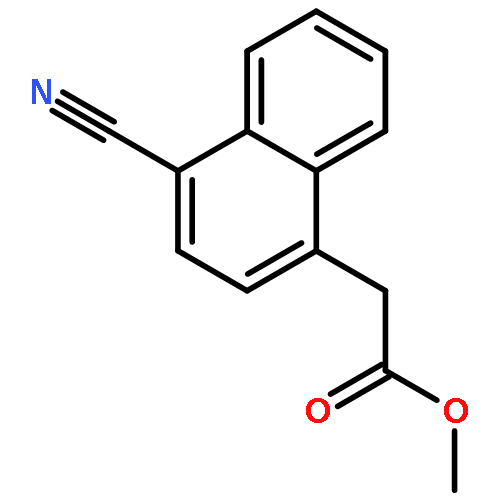
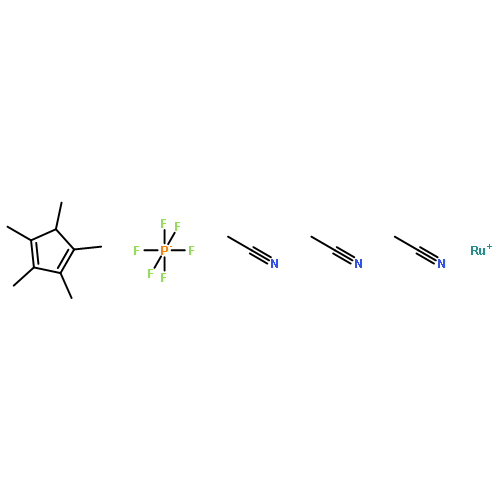
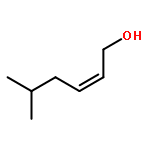
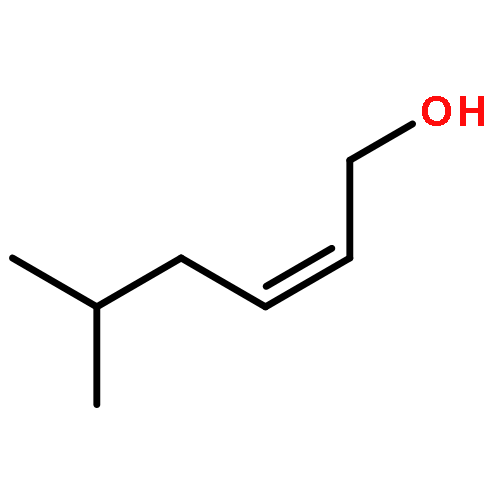
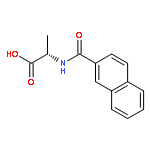
![5(2H)-Isoxazolone, 4-[(4-methoxyphenyl)methyl]-3-phenyl-](http://img.cochemist.com/ccimg/89200/89114-11-4.png)
![5(2H)-Isoxazolone, 4-[(4-methoxyphenyl)methyl]-3-phenyl-](http://img.cochemist.com/ccimg/89200/89114-11-4_b.png)
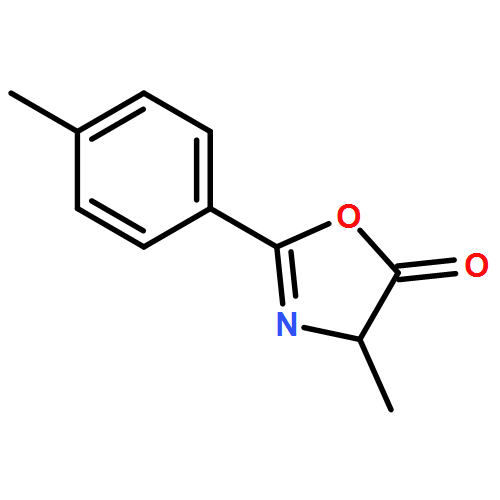
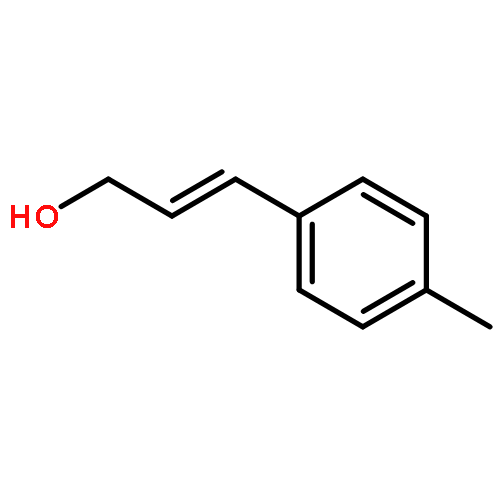
![5(2H)-ISOXAZOLONE, 4-[(4-METHYLPHENYL)METHYL]-3-PHENYL-](http://img.cochemist.com/ccimg/89200/89114-10-3.png)
![5(2H)-ISOXAZOLONE, 4-[(4-METHYLPHENYL)METHYL]-3-PHENYL-](http://img.cochemist.com/ccimg/89200/89114-10-3_b.png)
![(2s)-2-[tert-butyl(dimethyl)silyl]oxypropanal](http://img.cochemist.com/ccimg/87800/87727-28-4.png)
![(2s)-2-[tert-butyl(dimethyl)silyl]oxypropanal](http://img.cochemist.com/ccimg/87800/87727-28-4_b.png)
![Acetic acid, [[(4-methylphenyl)sulfonyl]imino]-, ethyl ester](http://img.cochemist.com/ccimg/83700/83670-53-5.png)
![Acetic acid, [[(4-methylphenyl)sulfonyl]imino]-, ethyl ester](http://img.cochemist.com/ccimg/83700/83670-53-5_b.png)
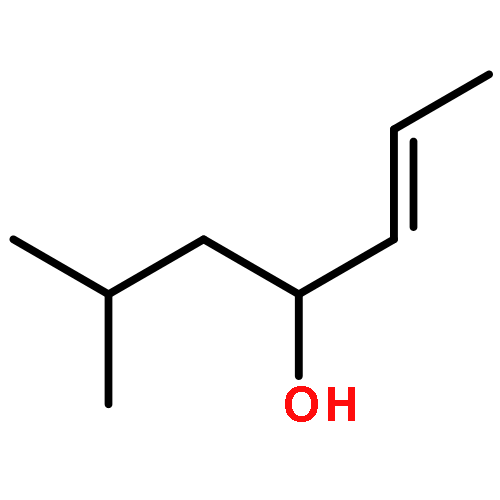
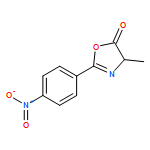
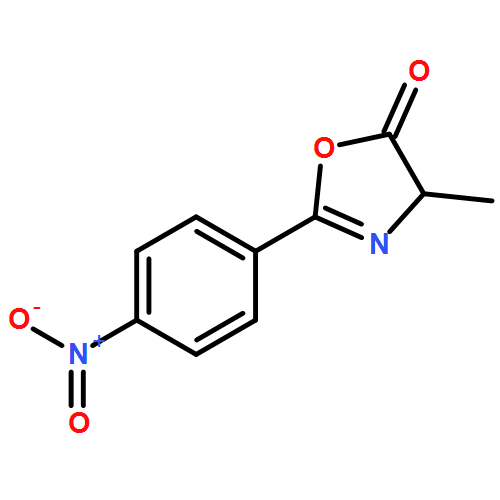
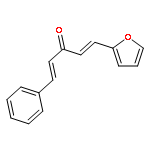
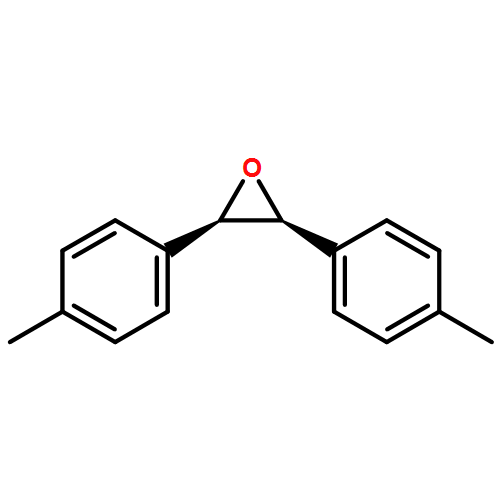
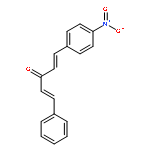
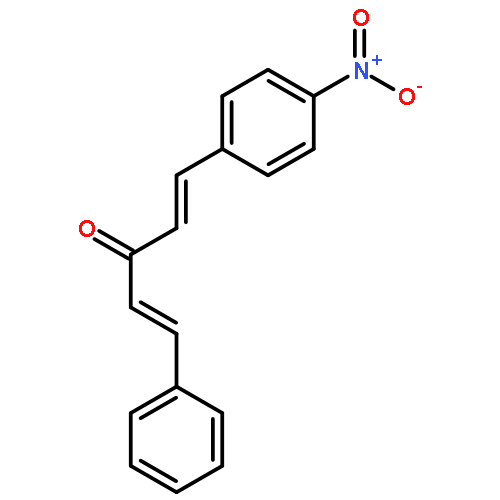
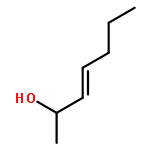
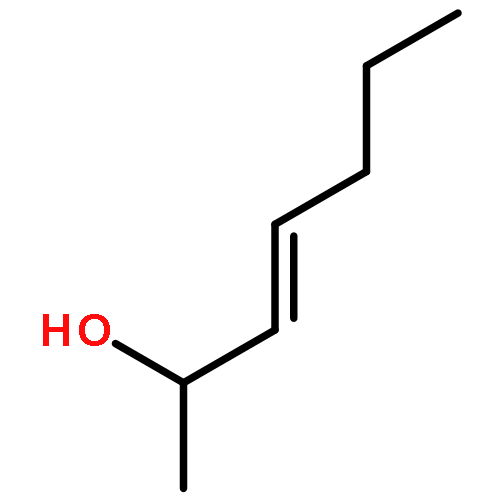
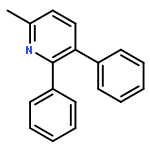
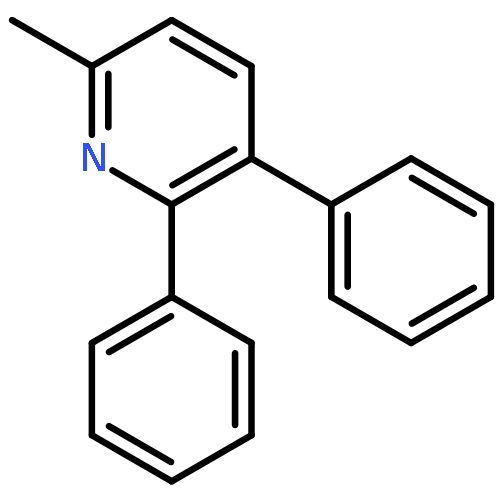
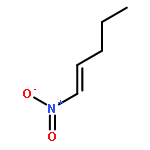
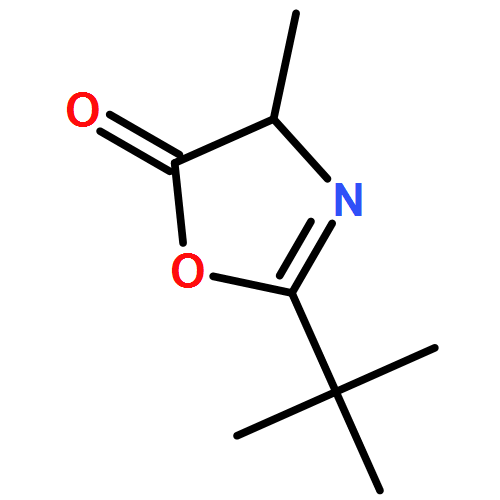
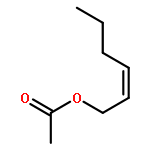
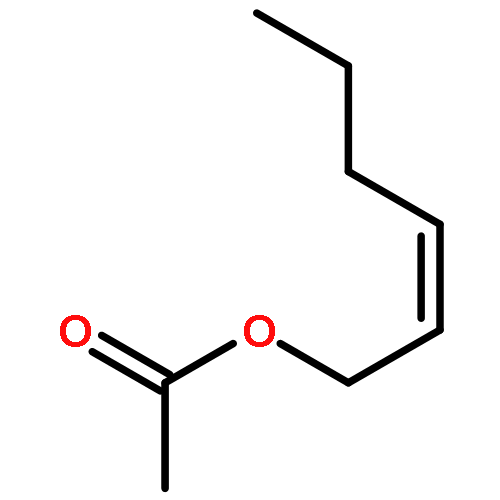
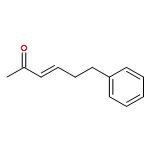
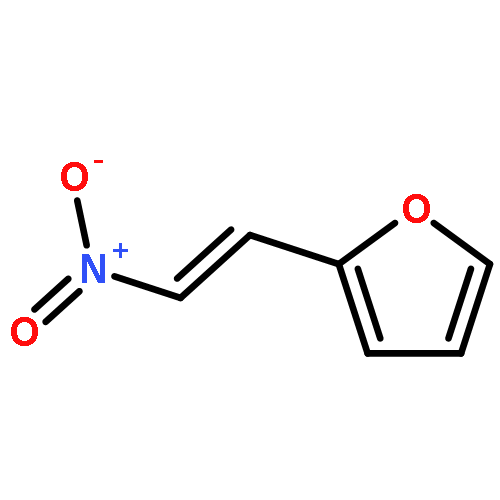
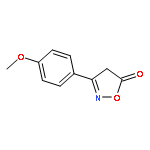
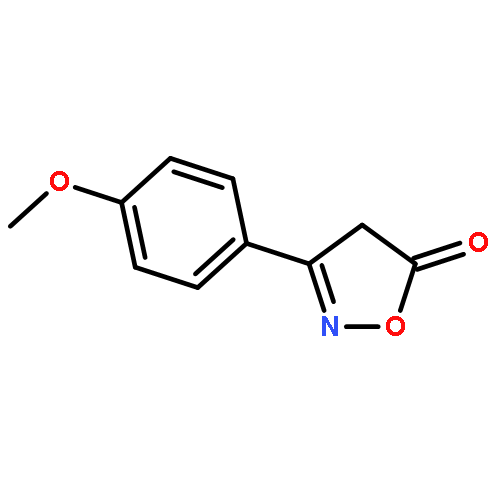
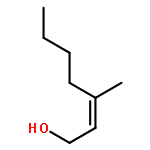
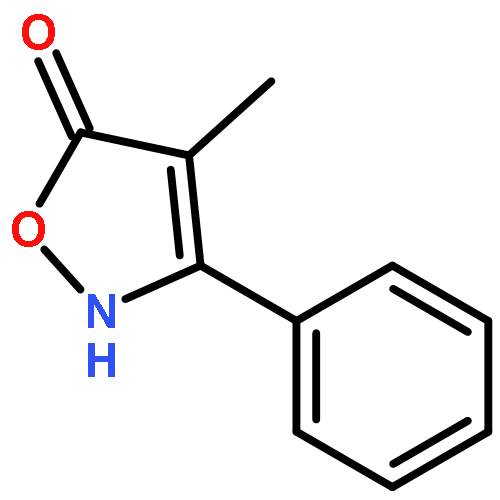
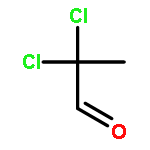
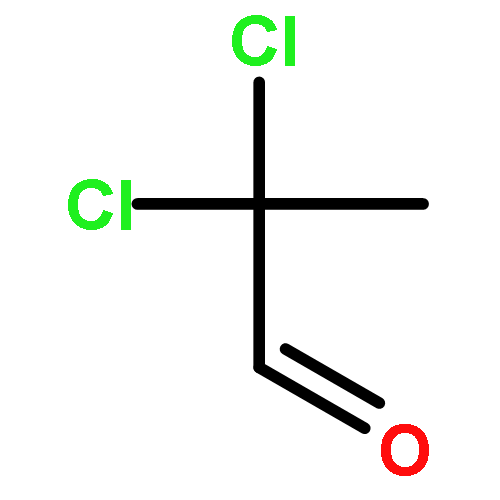
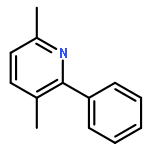
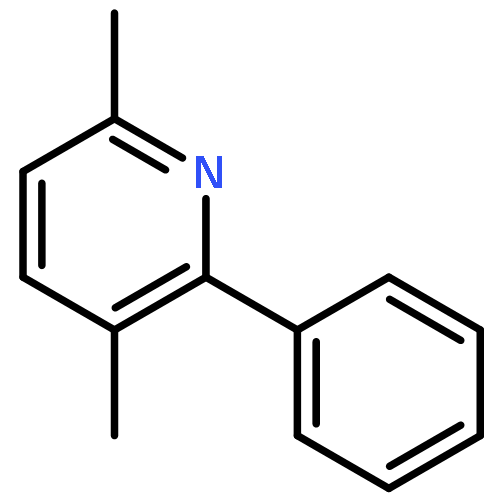
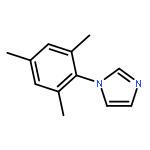
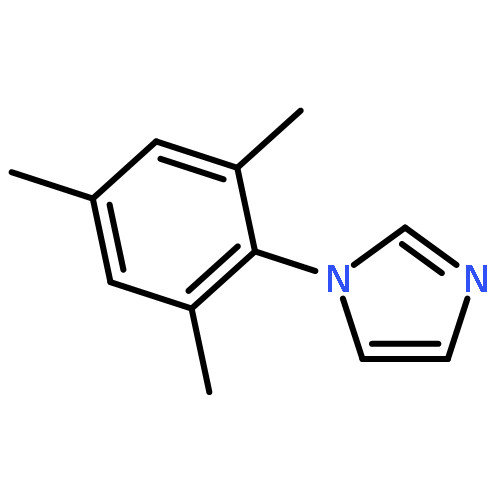
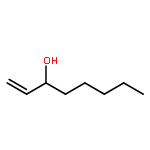
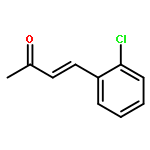
![1-chloro-2-[(e)-2-nitrovinyl]benzene](http://img.cochemist.com/ccimg/22600/22568-07-6.png)
![1-chloro-2-[(e)-2-nitrovinyl]benzene](http://img.cochemist.com/ccimg/22600/22568-07-6_b.png)
![Bicyclo[2.2.1]hept-5-ene-2-methanol, (1R,2R,4R)-rel-](/data/chemimg/1745500/15507-06-9.png)
![Bicyclo[2.2.1]hept-5-ene-2-methanol, (1R,2R,4R)-rel-](/data/chemimg/1745500/15507-06-9_b.png)


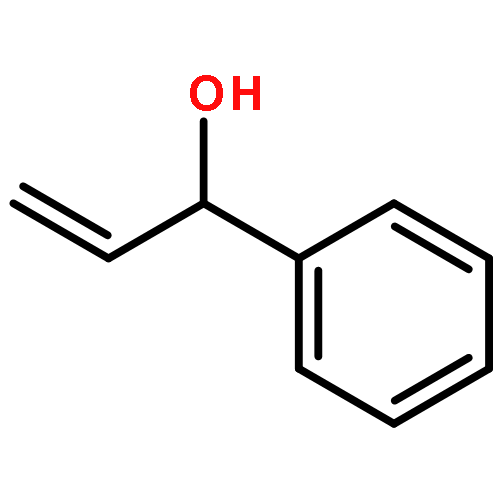
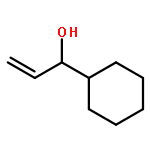
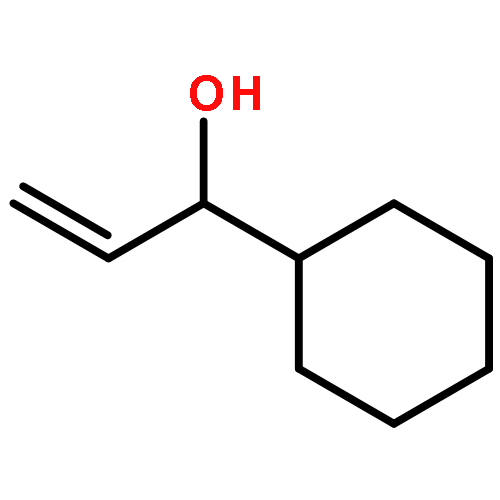
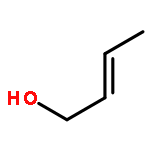
![1,1'-(1,6-HEXANEDIYL)BIS(3-BENZYL-1-AZONIABICYCLO[2.2.2]OCTANE) D<WBR />ICHLORIDE](http://img.cochemist.com/ccimg/4000/3913-80-2.png)
![1,1'-(1,6-HEXANEDIYL)BIS(3-BENZYL-1-AZONIABICYCLO[2.2.2]OCTANE) D<WBR />ICHLORIDE](http://img.cochemist.com/ccimg/4000/3913-80-2_b.png)
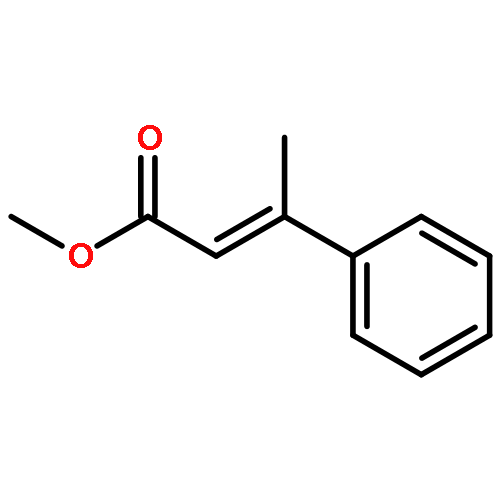
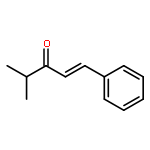
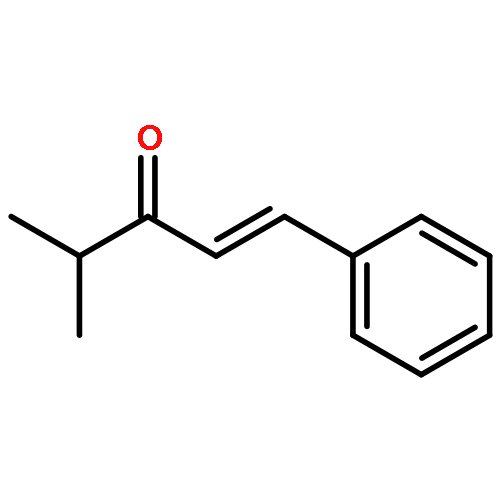
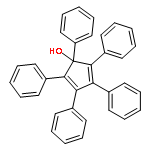
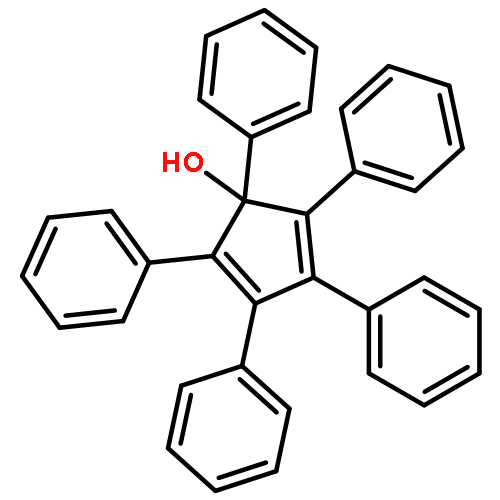


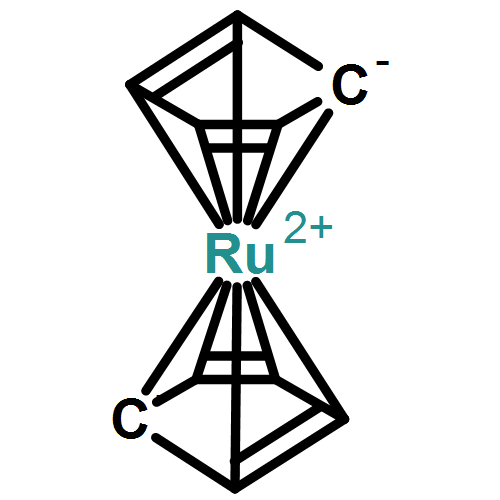
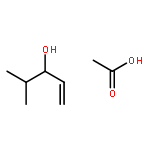
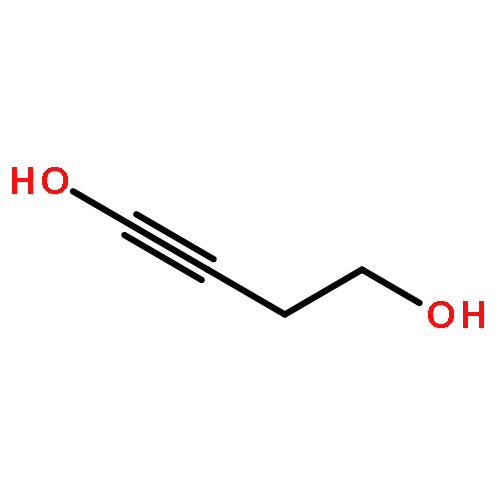
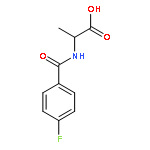
![2,2'-[oxybis(ethane-2,1-diyloxy)]bis[2-methylpropane]](http://img.cochemist.com/ccimg/200/107-79-9.png)
![2,2'-[oxybis(ethane-2,1-diyloxy)]bis[2-methylpropane]](http://img.cochemist.com/ccimg/200/107-79-9_b.png)




![Benzenesulfonamide, N-[(1S)-1-ethenylbutyl]-4-methyl-](http://img.cochemist.com/ccimg/869800/869740-62-5.png)
![Benzenesulfonamide, N-[(1S)-1-ethenylbutyl]-4-methyl-](http://img.cochemist.com/ccimg/869800/869740-62-5_b.png)
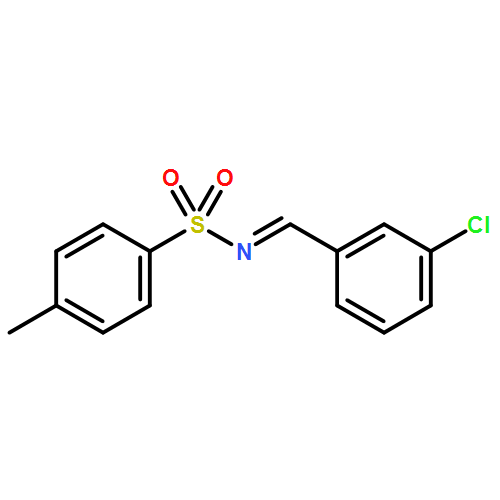
![Benzenesulfonamide, N-[(R)-(4-chlorophenyl)phenylmethyl]-4-nitro-](http://img.cochemist.com/ccimg/840600/840529-66-0.png)
![Benzenesulfonamide, N-[(R)-(4-chlorophenyl)phenylmethyl]-4-nitro-](http://img.cochemist.com/ccimg/840600/840529-66-0_b.png)
![BENZENESULFONAMIDE, N-[(R)-(4-METHOXYPHENYL)PHENYLMETHYL]-4-METHYL-](http://img.cochemist.com/ccimg/797000/796966-22-8.png)
![BENZENESULFONAMIDE, N-[(R)-(4-METHOXYPHENYL)PHENYLMETHYL]-4-METHYL-](http://img.cochemist.com/ccimg/797000/796966-22-8_b.png)
![Benzenesulfonamide, N-[(R)-(4-chlorophenyl)phenylmethyl]-4-methyl-](http://img.cochemist.com/ccimg/797000/796966-21-7.png)
![Benzenesulfonamide, N-[(R)-(4-chlorophenyl)phenylmethyl]-4-methyl-](http://img.cochemist.com/ccimg/797000/796966-21-7_b.png)
![BENZENESULFONAMIDE, N-[(S)-(4-METHOXYPHENYL)PHENYLMETHYL]-4-METHYL-](http://img.cochemist.com/ccimg/797000/796966-18-2.png)
![BENZENESULFONAMIDE, N-[(S)-(4-METHOXYPHENYL)PHENYLMETHYL]-4-METHYL-](http://img.cochemist.com/ccimg/797000/796966-18-2_b.png)
![BENZENESULFONAMIDE, 4-METHYL-N-[(1S)-2-METHYL-1-PHENYLPROPYL]-](http://img.cochemist.com/ccimg/627600/627503-87-1.png)
![BENZENESULFONAMIDE, 4-METHYL-N-[(1S)-2-METHYL-1-PHENYLPROPYL]-](http://img.cochemist.com/ccimg/627600/627503-87-1_b.png)
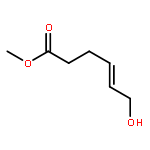
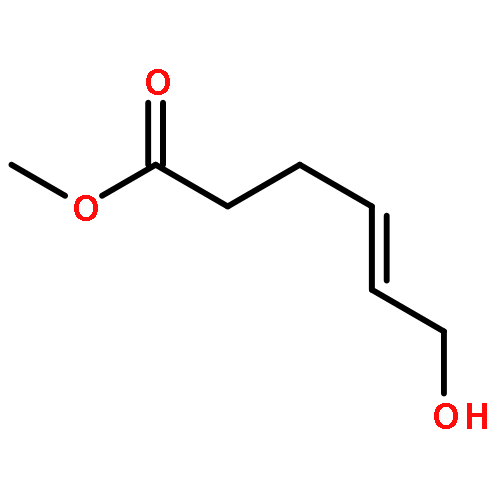
![Acetamide, N-[(1R)-1-ethenylbutyl]-2,2,2-trifluoro-N-(4-methoxyphenyl)-](http://img.cochemist.com/ccimg/562900/562870-84-2.png)
![Acetamide, N-[(1R)-1-ethenylbutyl]-2,2,2-trifluoro-N-(4-methoxyphenyl)-](http://img.cochemist.com/ccimg/562900/562870-84-2_b.png)
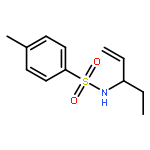

![BENZENESULFONAMIDE, 4-METHYL-N-[(1R)-1-METHYL-2-PROPENYL]-](http://img.cochemist.com/ccimg/461700/461677-19-0.png)
![BENZENESULFONAMIDE, 4-METHYL-N-[(1R)-1-METHYL-2-PROPENYL]-](http://img.cochemist.com/ccimg/461700/461677-19-0_b.png)
![Benzenesulfonamide, N-[(1R)-1-ethenylhexyl]-4-methyl-](http://img.cochemist.com/ccimg/397400/397304-51-7.png)
![Benzenesulfonamide, N-[(1R)-1-ethenylhexyl]-4-methyl-](http://img.cochemist.com/ccimg/397400/397304-51-7_b.png)




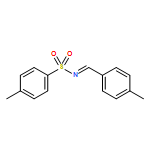
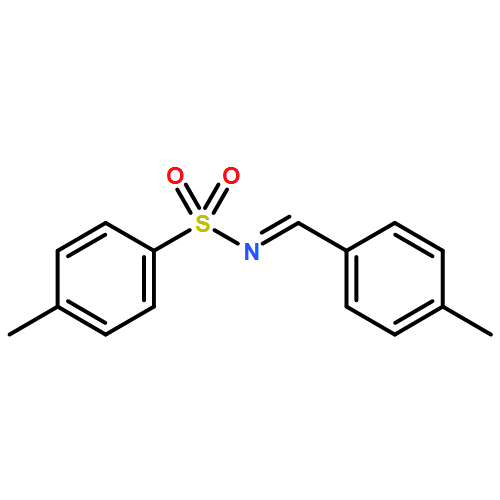
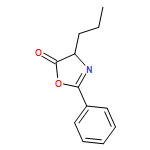
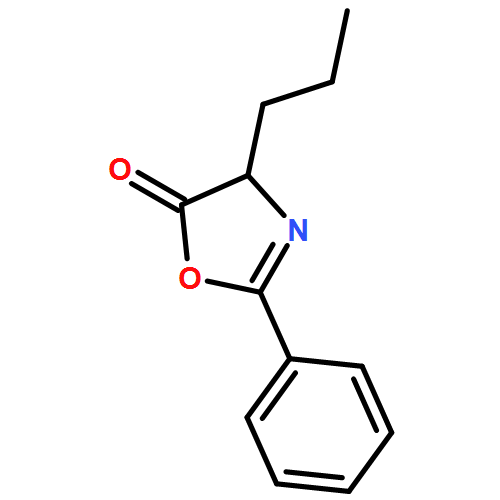

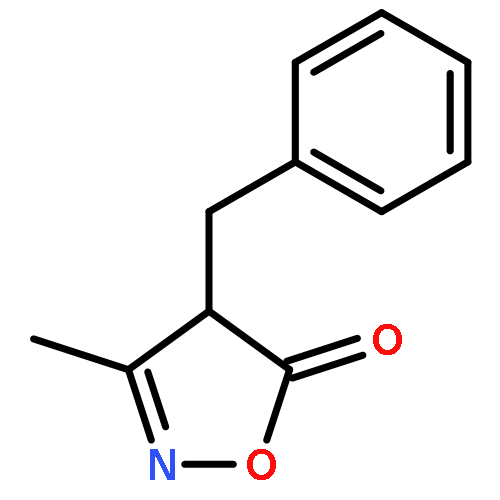
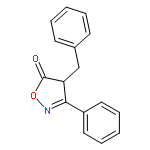
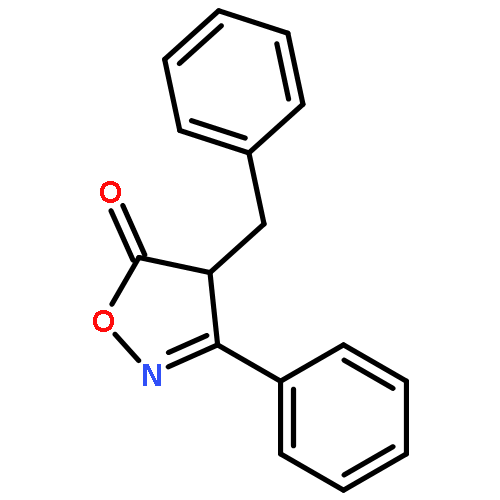
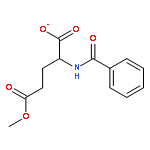

![Benzene, 1-methoxy-3-[(1E)-2-nitroethenyl]-](http://img.cochemist.com/ccimg/55500/55446-68-9.png)
![Benzene, 1-methoxy-3-[(1E)-2-nitroethenyl]-](http://img.cochemist.com/ccimg/55500/55446-68-9_b.png)

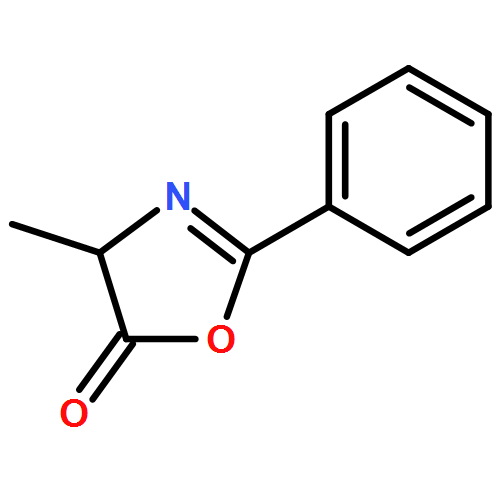
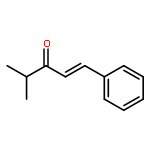
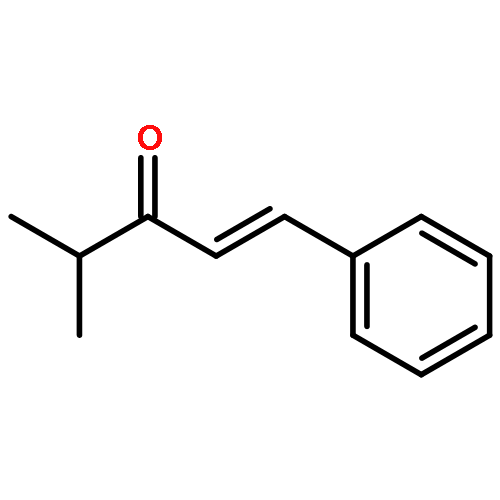
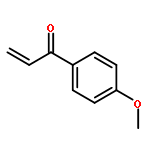


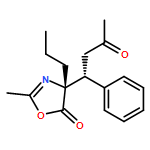
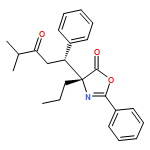
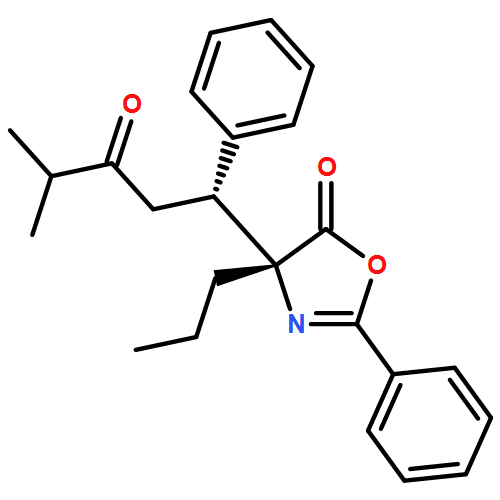
![5(4H)-Oxazolone, 4-[(1R)-1-(1-methylethyl)-3-oxobutyl]-2-phenyl-4-propyl-, (4R)-](/data/chemimg/3721700/1242985-42-7.png)
![5(4H)-Oxazolone, 4-[(1R)-1-(1-methylethyl)-3-oxobutyl]-2-phenyl-4-propyl-, (4R)-](/data/chemimg/3721700/1242985-42-7_b.png)
![5(4H)-Oxazolone, 4-[(1R)-1-(4-nitrophenyl)-3-oxobutyl]-2-phenyl-4-propyl-, (4R)-](/data/chemimg/3721700/1242985-39-2.png)
![5(4H)-Oxazolone, 4-[(1R)-1-(4-nitrophenyl)-3-oxobutyl]-2-phenyl-4-propyl-, (4R)-](/data/chemimg/3721700/1242985-39-2_b.png)
![5(4H)-Oxazolone, 4-[(1R)-1-(2-chlorophenyl)-3-oxobutyl]-2-phenyl-4-propyl-, (4R)-](/data/chemimg/3721700/1242985-38-1.png)
![5(4H)-Oxazolone, 4-[(1R)-1-(2-chlorophenyl)-3-oxobutyl]-2-phenyl-4-propyl-, (4R)-](/data/chemimg/3721700/1242985-38-1_b.png)
![5(4H)-Oxazolone, 4-[(1R)-1-(4-methoxyphenyl)-3-oxobutyl]-2-phenyl-4-propyl-, (4R)-](/data/chemimg/3721700/1242985-36-9.png)
![5(4H)-Oxazolone, 4-[(1R)-1-(4-methoxyphenyl)-3-oxobutyl]-2-phenyl-4-propyl-, (4R)-](/data/chemimg/3721700/1242985-36-9_b.png)
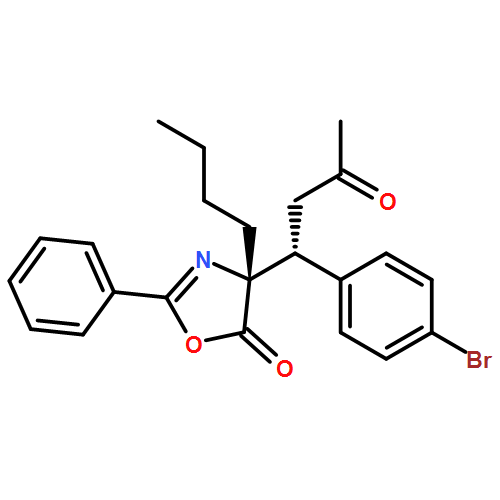
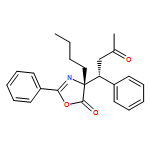
![5(4H)-Oxazolone, 4-[(1R)-1-(4-bromophenyl)-3-oxobutyl]-4-ethyl-2-phenyl-, (4R)-](/data/chemimg/3721700/1242985-31-4.png)
![5(4H)-Oxazolone, 4-[(1R)-1-(4-bromophenyl)-3-oxobutyl]-4-ethyl-2-phenyl-, (4R)-](/data/chemimg/3721700/1242985-31-4_b.png)
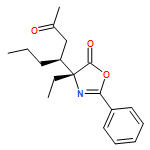
![5(4H)-Oxazolone, 4-ethyl-4-[(1R)-3-oxo-1-phenylbutyl]-2-phenyl-, (4R)-](/data/chemimg/3721600/1242985-26-7.png)
![5(4H)-Oxazolone, 4-ethyl-4-[(1R)-3-oxo-1-phenylbutyl]-2-phenyl-, (4R)-](/data/chemimg/3721600/1242985-26-7_b.png)
![BENZENAMINE, N-[(1S)-1-ETHENYLBUTYL]-4-METHOXY-](http://img.cochemist.com/ccimg/562900/562870-91-1.png)
![BENZENAMINE, N-[(1S)-1-ETHENYLBUTYL]-4-METHOXY-](http://img.cochemist.com/ccimg/562900/562870-91-1_b.png)
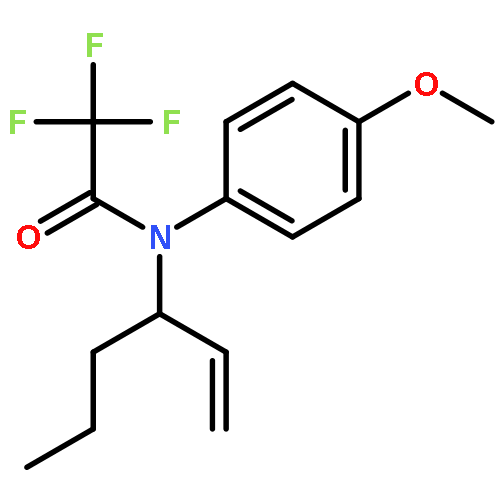
![Benzenesulfonamide, N-[(1R)-1-ethenylbutyl]-4-methyl-](http://img.cochemist.com/ccimg/369400/369354-95-0.png)
![Benzenesulfonamide, N-[(1R)-1-ethenylbutyl]-4-methyl-](http://img.cochemist.com/ccimg/369400/369354-95-0_b.png)
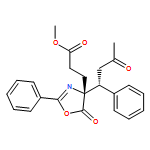
![5(4H)-Oxazolone, 4-[(1R)-3-oxo-1-phenylbutyl]-2-phenyl-4-(phenylmethyl)-, (4R)-](/data/chemimg/3721500/1242985-44-9.png)
![5(4H)-Oxazolone, 4-[(1R)-3-oxo-1-phenylbutyl]-2-phenyl-4-(phenylmethyl)-, (4R)-](/data/chemimg/3721500/1242985-44-9_b.png)
![5(4H)-Oxazolone, 4-[(1R)-3-oxo-1,3-diphenylpropyl]-2-phenyl-4-propyl-, (4R)-](/data/chemimg/3721600/1242985-41-6.png)
![5(4H)-Oxazolone, 4-[(1R)-3-oxo-1,3-diphenylpropyl]-2-phenyl-4-propyl-, (4R)-](/data/chemimg/3721600/1242985-41-6_b.png)
![5(4H)-Oxazolone, 4-[(1S)-1-(2-furanyl)-3-oxobutyl]-2-phenyl-4-propyl-, (4R)-](/data/chemimg/3721600/1242985-40-5.png)
![5(4H)-Oxazolone, 4-[(1S)-1-(2-furanyl)-3-oxobutyl]-2-phenyl-4-propyl-, (4R)-](/data/chemimg/3721600/1242985-40-5_b.png)
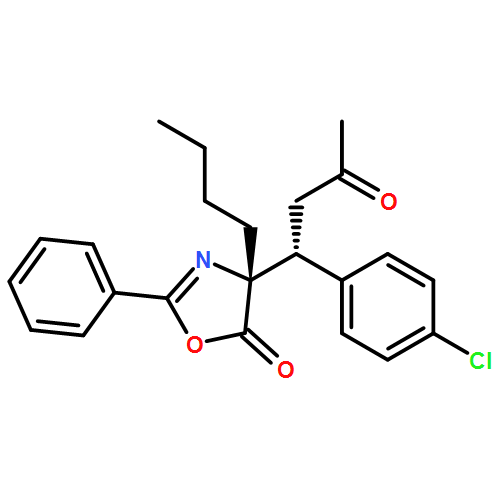
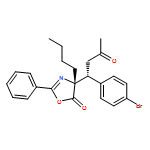
![5(4H)-Oxazolone, 4-[(1R)-1-(4-chlorophenyl)-3-oxobutyl]-2-phenyl-4-propyl-, (4R)-](/data/chemimg/3721600/1242985-37-0.png)
![5(4H)-Oxazolone, 4-[(1R)-1-(4-chlorophenyl)-3-oxobutyl]-2-phenyl-4-propyl-, (4R)-](/data/chemimg/3721600/1242985-37-0_b.png)
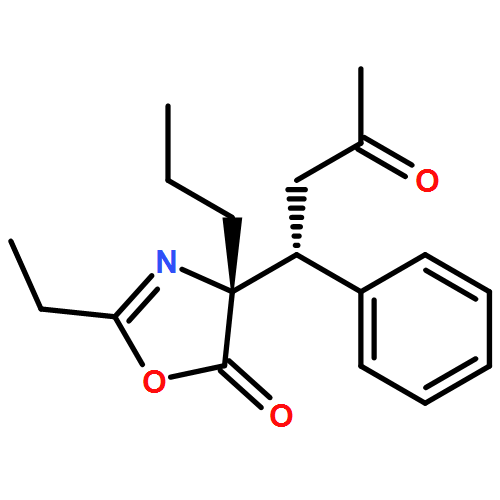
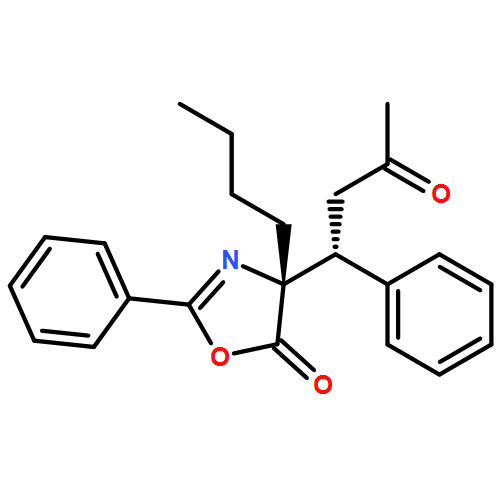
![5(4H)-Oxazolone, 4-[(1R)-3-oxo-1-phenylbutyl]-2-phenyl-4-propyl-, (4R)-](/data/chemimg/3721500/1242985-35-8.png)
![5(4H)-Oxazolone, 4-[(1R)-3-oxo-1-phenylbutyl]-2-phenyl-4-propyl-, (4R)-](/data/chemimg/3721500/1242985-35-8_b.png)
![5(4H)-Oxazolone, 4-methyl-4-[(1R)-3-oxo-1-phenylbutyl]-2-phenyl-, (4R)-](/data/chemimg/3721500/1242985-18-7.png)
![5(4H)-Oxazolone, 4-methyl-4-[(1R)-3-oxo-1-phenylbutyl]-2-phenyl-, (4R)-](/data/chemimg/3721500/1242985-18-7_b.png)