Co-reporter:Yasuhiro Arikawa;Takuo Nakamura;Takefumi Higashi;Shinnosuke Horiuchi;Eri Sakuda;Keisuke Umakoshi
European Journal of Inorganic Chemistry 2017 Volume 2017(Issue 5) pp:881-884
Publication Date(Web):2017/02/03
DOI:10.1002/ejic.201601324
Thiocyanate linkage isomers and two insertion complexes were prepared from a methoxido–ruthenium complex bearing a 2,6-bis(3-tert-butylimidazol-2-ylidene)pyridine (CNC) and a bipyridine ligand. In the mixture of the linkage isomers obtained from the substitution reaction, a linear N-bound and a bent S-bound isomer were crystallographically determined, and the equilibrium between them at elevated temperature was revealed. On the other hand, in reactions with carbon disulfide (CS2) and phenyl isothiocyanate (PhNCS), the S=C bond was inserted into the Ru–OMe bond, and the resulting ligands are bound to the ruthenium center by the sulfur atom.
Co-reporter:Takuo Nakamura, Shinji Ogushi, Yasuhiro Arikawa, Keisuke Umakoshi
Journal of Organometallic Chemistry 2016 Volume 803() pp:67-72
Publication Date(Web):1 February 2016
DOI:10.1016/j.jorganchem.2015.12.006
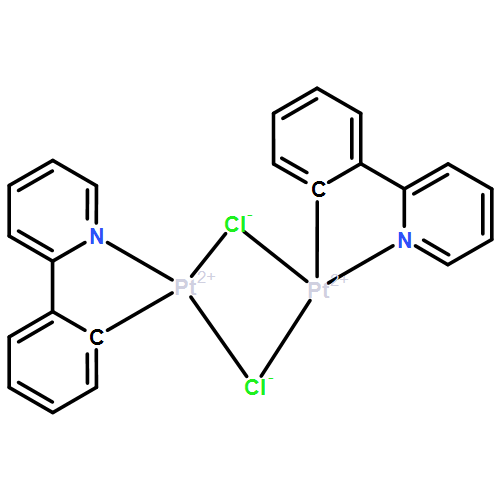
•A silver complex bearing 2,6-bis(3-tert-butylimidazol-2-ylidene)pyridine ligands.•The carbene transfer reactions resulted in a series of coinage metal complexes.•The palladium complexes were also prepared from the carbene transfer reactions.A silver complex bearing 2,6-bis(3-tert-butylimidazol-2-ylidene)pyridine (CNC) ligands was easily prepared from the reaction of the CNC ligand precursor with Ag2O. The CNC ligand contains relatively bulky substituents, tert-butyl, on nitrogen atoms. Use of the silver complex as a carbene transfer reagent gave rise to a copper and a gold complex, completing a series of coinage metal complexes [M2(CNC)2](BF4)2 (M = Cu, Ag, Au). In all the coinage metal complexes, the X-ray crystallographic analyses showed that the two CNC ligand strands are intertwined each other and surround the metal ions in a double helical fashion. Moreover, the carbene transfer reactions afforded a monomeric palladium complex [Pd(CNC)Cl]BF4 and a CNC-bridged dipalladium complex [{Pd(η3-C3H5)Cl}2(μ-CNC)]. Facile chloride ligand abstraction from the dipalladium complex yielded a chlorido-bridged dipalladium complex [{Pd(η3-C3H5)}2(μ-CNC)(μ-Cl)]BF4.
Co-reporter:Tatsuya Suzuki; Hiromasa Tanaka; Yoshihito Shiota; P. K. Sajith; Yasuhiro Arikawa;Kazunari Yoshizawa
Inorganic Chemistry 2015 Volume 54(Issue 15) pp:7181-7191
Publication Date(Web):July 17, 2015
DOI:10.1021/acs.inorgchem.5b00394
Density-functional-theory (DFT) calculations are performed for the proposal of a plausible mechanism on the reduction of NO to N2O by a dinuclear ruthenium complex, reported by Arikawa and co-workers [J. Am. Chem. Soc. 2007, 129, 14160]. On the basis of the experimental fact that the reduction proceeds under strongly acidic conditions, the role of protons in the mechanistic pathways is investigated with model complexes, where one or two NO ligands are protonated. The reaction mechanism of the NO reduction is partitioned into three steps: reorientation of N2O2 (cis-NO dimer), O—N bond cleavage, and N2O elimination. A key finding is that the protonation of the NO ligand(s) significantly reduces the activation barrier in the rate-determining reorientation step. The activation energy of 43.1 kcal/mol calculated for the proton-free model is reduced to 30.2 and 17.6 kcal/mol for the mono- and diprotonated models, respectively. The protonation induces the electron transfer from the Ru(II)Ru(II) core to the O═N—N═O moiety to give a Ru(III)Ru(III) core and a hyponitrite (O—N═N—O)2– species. The formation of the hyponitrite species provides an alternative pathway for the N2O2 reorientation, resulting in the lower activation energies in the presence of proton(s). The protonation also has a marginal effect on the O—N bond cleavage and the N2O elimination steps. Our calculations reveal a remarkable role of protons in the NO reduction via N2O formation and provide new insights into the mechanism of NO reduction catalyzed by metalloenzymes such as nitric oxide reductase (NOR) that contains a diiron active site.
Co-reporter:Yasuhiro Arikawa, Takuo Nakamura, Shinji Ogushi, Kazushige Eguchi and Keisuke Umakoshi
Dalton Transactions 2015 vol. 44(Issue 12) pp:5303-5305
Publication Date(Web):12 Feb 2015
DOI:10.1039/C5DT00476D
A methylcarbonato ruthenium complex was prepared by capture of CO2 from air using the (CNC)(bpy)Ru scaffold. The methylcarbonato complex was relatively inert to decarboxylation. Treatments with methylating reagents released dimethylcarbonate.
Co-reporter:Yasuhiro Arikawa, Soseki Yamaguchi, Yuji Otsubo, Masayoshi Onishi, and Keisuke Umakoshi
Organometallics 2015 Volume 34(Issue 6) pp:1056-1061
Publication Date(Web):March 3, 2015
DOI:10.1021/om5012769
Nitrosation of anilines at the ortho position was found to proceed on a ruthenium hydridotris(pyrazolyl)borato (Tp) complex. Reactions of [TpRuCl2(NO)] (1) with primary anilines 4-NH2C6H4R (R = tBu, H) in the presence of excess Et3N in CH2Cl2 gave amine-chelated nitrosoarene complexes [TpRuCl{N(═O)–C6H3R–NH2-κ2N,N}] (R = tBu (2a), H (2b)). Use of 2,4,6-trimethylaniline afforded an aryldiazenido complex [TpRuCl2{NNC6H2(Me)3}] (3) without forming the nitrosation product because of the introduction of the Me substituents at the ortho positions. On the other hand, in the case of secondary amines (N-methylanilines 4-NH(Me)C6H4R (R = tBu, H)), similar reactions gave amine-chelated nitroso complexes [TpRuCl{N(═O)–C6H3R–NHMe-κ2N,N}] (R = tBu (4a), H (4b)) and imine-chelated nitroso complexes [TpRuCl{N(═O)–C6H3R–N═CH2-κ2N,N}] (R = tBu (5a), H (5b)). Conversion of 4b into 5b by O2 was disclosed by 1H NMR monitoring. Moreover, oxidative reaction of 2a afforded an amide-chelated nitroso complex [TpRuCl{N(═O)–C6H3(tBu)–NH-κ2N,N}] (6a) through one-proton release from the NH2 group.
Co-reporter:Yasuhiro Arikawa, Soseki Yamaguchi, Ryohei Haige, Eriko Oshiro, Keisuke Umakoshi, Masayoshi Onishi
Journal of Organometallic Chemistry 2014 Volume 755() pp:12-15
Publication Date(Web):1 April 2014
DOI:10.1016/j.jorganchem.2013.12.054
•Mono- and dimethyl(nitrosyl)ruthenium complexes supported by a Tp ligand were prepared.•Use of dimethylzinc as alkylating reagents led to isolation of methyl complexes in good yields.•The dimethyl complex is thermally robust.•Protonolysis of the dimethyl complex with triflic acid resulted in a monomethyl triflato complex.Reaction of [TpRuCl2(NO)] (1) (Tp = HB(pyrazol-1-yl)3) with dimethylzinc, Zn(Me)2, gave rise to a dimethyl complex [TpRu(Me)2(NO)] (2) and a monomethyl complex [TpRuCl(Me)(NO)] (3) in good yields, while the use of a Grignard reagent, MeMgCl, as the alkylating agents led to isolation of 2 in low yield. Complexes 2 and 3 were confirmed by single-crystal X-ray diffraction analyses. Treatment of 2 with triflic acid, CF3SO3H, afforded a triflato complex [TpRu(Me){OS(O)2CF3}(NO)] (4).Reaction of [TpRuCl2(NO)] (1) (Tp = HB(pyrazol-1-yl)3) with dimethylzinc, Zn(Me)2, gave rise to a dimethyl complex [TpRu(Me)2(NO)] (2) and a monomethyl complex [TpRuCl(Me)(NO)] (3) in good yields. Treatment of 2 with triflic acid, CF3SO3H, afforded a triflato complex [TpRu(Me){OS(O)2CF3}(NO)] (4).
Co-reporter:Yasuhiro Arikawa, Ayumi Ikeda, Naoki Matsumoto and Keisuke Umakoshi
Dalton Transactions 2013 vol. 42(Issue 32) pp:11626-11631
Publication Date(Web):14 Jun 2013
DOI:10.1039/C3DT51319J
A cationic mononitrosyl dinuclear ruthenium complex was prepared by removing one NO ligand of a dicationic dinitrosyl ruthenium complex using NaN3. Reduction and oxidation reactions of the mononitrosyl complex led to the isolation of a neutral nitrosyl-bridged complex and a dicationic mononitrosyl complex, respectively, as expected from the cyclic voltammogram. According to the electron count, their reactions with a second NO molecule resulted in an N–N coupling complex from the nitrosyl-bridged complex and the dicationic dinitrosyl complex from the dicationic mononitrosyl complex.
Co-reporter:Yasuhiro Arikawa, Masayoshi Onishi
Coordination Chemistry Reviews 2012 Volume 256(5–8) pp:468-478
Publication Date(Web):March–April 2012
DOI:10.1016/j.ccr.2011.10.023
Nitric oxide reductase (NOR) type reactions (2NO + 2e− + 2H+ → N2O + H2O) on transition metal complexes not involving NO disproportionation (3NO → N2O + NO2) are reviewed. The former has little reported, although the latter is very common reaction. The formation of N2O indicates that N–N coupling of two NO molecules is an essential step. A few examples of N–N coupling on transition metal complexes have been structurally characterized, including several examples of hyponitrite (O–NN–O)2− complexes and only one diruthenium complex bearing neutral (ON–NO) binding mode. Protonation or heating their complexes led to elimination of N2O. In the examination of the NOR-type reaction, only a few functional model complexes for the active site of the metalloenzyme have been developed. These complexes also showed NOR activity. Finally, an NO reduction cycle in the diruthenium system is described.Graphical abstractHighlights► NOR-type reactions (2NO + 2e− + 2H+ → N2O + H2O) on transition metal complexes. ► Key step is N–N coupling of 2 NO molecules on transition metal complexes. ► Several structurally characterized hyponitrite (O–NN–O)2− complexes. ► Only one diruthenium complex bearing neutral (ON–NO) binding mode. ► A few biomimetic model complexes showed the NOR-type reaction.
Co-reporter:Yasuhiro Arikawa, Naoki Matsumoto, Taiki Asayama, Keisuke Umakoshi and Masayoshi Onishi
Dalton Transactions 2011 vol. 40(Issue 10) pp:2148-2150
Publication Date(Web):13 Oct 2010
DOI:10.1039/C0DT01002B
The hydroxido-bridged dinuclear ruthenium complex 4, which is supported by Tp ligands, has been prepared from protonation of the oxido-bridged dinuclear ruthenium complex 3. Additional protonation of 4, affording the aqua-bridged dinuclear ruthenium complex 5in situ, and subsequent treatment with NO gave rise to the dicationic dinitrosyl complex 2. These indicate completion of the NO reduction cycle on the dinuclear ruthenium complex.
Co-reporter:Yasuhiro Arikawa, Hayato Yamasaki, Mamoru Yamaguchi, Keisuke Umakoshi and Masayoshi Onishi
Organometallics 2009 Volume 28(Issue 18) pp:5587-5589
Publication Date(Web):August 26, 2009
DOI:10.1021/om900576h
Treatment of a ruthenium vinylidene complex bearing a bent-type NO ligand, RuCl(NO)(PPh3)2{═C═CH(C6H4Me)} (1), with protic acids gave rise to the ruthenium vinyl complexes RuCl(X){(Z)-CH═CH(C6H4Me)}(PPh3)2(NO) (X = Cl (2a), O2CCF3 (2b), FBF3 (2c)) with concomitant transformation to linear-type NO. This indicates unusal protonation at the vinylidene α-carbon and anion-part coordination of the protic acids to the ruthenium atom at the trans position to the resulting vinyl group. The coordinated BF4 moiety of 2c was easily replaced to be a counteranion by MeCN solvent molecules, yielding [RuCl{(Z)-CH═CH(C6H4Me)}(NCMe)(PPh3)2(NO)]BF4 (3).
Co-reporter:Yasuhiro Arikawa, Taiki Asayama, Chie Tanaka, Shin-ya Tashita, Misako Tsuji, Kenta Ikeda, Keisuke Umakoshi and Masayoshi Onishi
Organometallics 2008 Volume 27(Issue 6) pp:1227-1233
Publication Date(Web):February 27, 2008
DOI:10.1021/om701028r
Nitrosylruthenium arylbutadiynyl complexes having a Tp ligand (Tp = BH(pyrazol-1-yl)3) were prepared, and their reactivities toward PPh3 incorporation in the presence of HBF4·Et2O were described. The PPh3 incorporation of mono(arylbutadiynyl) complex TpRuCl(C≡C−C≡C−C6H4Me)(NO) (1) resulted in the β-phosphonioalkenyl complex (E)-[TpRuCl(CH═C(PPh3)−C≡C−C6H4Me)(NO)]BF4 (2·BF4), whereas when bis(arylbutadiynyl) TpRu(C≡C−C≡C−C6H4Me)2(NO) (3) was treated, mono- and bis(β-phosphonioalkenyl) complexes (E)-[TpRu(C≡C−C≡C−C6H4Me)(CH═C(PPh3)−C≡C−C6H4Me)(NO)]BF4 (4·BF4) and (E, E)-[TpRu(CH═C(PPh3)−C≡C−C6H4Me)2(NO)](BF4)2 {5·(BF4)2} were obtained depending on the reaction conditions. On the other hand, an unsymmetrically mixed (arylbutadiynyl)(3-hydroxyalkynyl) complex, TpRu(C≡C−C≡C−C6H4Me){C≡CCPh2(OH)}(NO) (6), was allowed to react with PPh3 in the presence of the protic acid to give the α-phosphonioallenyl [TpRu(C≡C−C≡C−C6H4Me){C(PPh3)═C═CPh2}(NO)]BF4 (7·BF4). Interestingly, thermal isomerization of 7·BF4 to a ruthena-2-PPh3-cyclobuta[b]naphthalene [TpRu{CH(PPh3)[3-Ph-8-(MeC6H4−C≡C)−C10H4]}(NO)]BF4 (8·BF4) was observed.
Co-reporter:Yasuhiro Arikawa, Naoki Matsumoto, Taiki Asayama, Keisuke Umakoshi and Masayoshi Onishi
Dalton Transactions 2011 - vol. 40(Issue 10) pp:NaN2150-2150
Publication Date(Web):2010/10/13
DOI:10.1039/C0DT01002B
The hydroxido-bridged dinuclear ruthenium complex 4, which is supported by Tp ligands, has been prepared from protonation of the oxido-bridged dinuclear ruthenium complex 3. Additional protonation of 4, affording the aqua-bridged dinuclear ruthenium complex 5in situ, and subsequent treatment with NO gave rise to the dicationic dinitrosyl complex 2. These indicate completion of the NO reduction cycle on the dinuclear ruthenium complex.
Co-reporter:Yasuhiro Arikawa, Takuo Nakamura, Shinji Ogushi, Kazushige Eguchi and Keisuke Umakoshi
Dalton Transactions 2015 - vol. 44(Issue 12) pp:NaN5305-5305
Publication Date(Web):2015/02/12
DOI:10.1039/C5DT00476D
A methylcarbonato ruthenium complex was prepared by capture of CO2 from air using the (CNC)(bpy)Ru scaffold. The methylcarbonato complex was relatively inert to decarboxylation. Treatments with methylating reagents released dimethylcarbonate.
Co-reporter:Yasuhiro Arikawa, Ayumi Ikeda, Naoki Matsumoto and Keisuke Umakoshi
Dalton Transactions 2013 - vol. 42(Issue 32) pp:NaN11631-11631
Publication Date(Web):2013/06/14
DOI:10.1039/C3DT51319J
A cationic mononitrosyl dinuclear ruthenium complex was prepared by removing one NO ligand of a dicationic dinitrosyl ruthenium complex using NaN3. Reduction and oxidation reactions of the mononitrosyl complex led to the isolation of a neutral nitrosyl-bridged complex and a dicationic mononitrosyl complex, respectively, as expected from the cyclic voltammogram. According to the electron count, their reactions with a second NO molecule resulted in an N–N coupling complex from the nitrosyl-bridged complex and the dicationic dinitrosyl complex from the dicationic mononitrosyl complex.

![Pentacyclo[19.3.1.13,7.19,13.115,19]octacosa-1(25),3,5,7(28),9,11,13(27),15,17,19(26),21,23-dodecaene-4,5,6,10,11,12,16,17,18,22,23,24-dodecol, 2,8,14,20-tetraundecyl-](/data/chemimg/102400/127203-21-8.png)
![Pentacyclo[19.3.1.13,7.19,13.115,19]octacosa-1(25),3,5,7(28),9,11,13(27),15,17,19(26),21,23-dodecaene-4,5,6,10,11,12,16,17,18,22,23,24-dodecol, 2,8,14,20-tetraundecyl-](/data/chemimg/102400/127203-21-8_b.png)