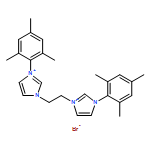
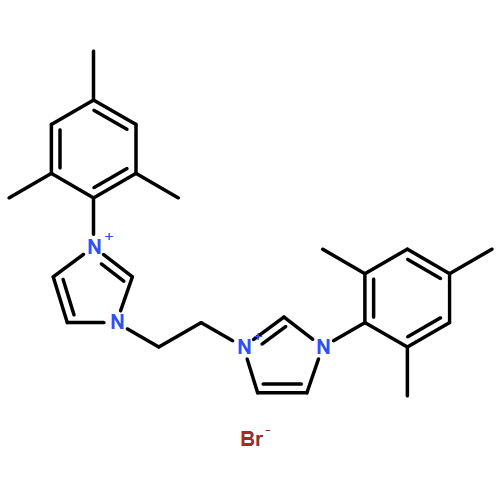
Co-reporter:Soraya N. Sluijter, Ties J. Korstanje, Jarl Ivar van der Vlugt, Cornelis J. Elsevier
Journal of Organometallic Chemistry 2017 Volume 845(Volume 845) pp:
Publication Date(Web):15 September 2017
DOI:10.1016/j.jorganchem.2017.01.003
•Synthesis of a novel bis(triazolyl)pyridine pincer ligand.•Coordination chemistry toward Ru(II).•Efficient hydrogenolysis of carboxylic esters.Transmetallation of newly designed lutidine-based CNC or CNN ligands L, featuring flanking 1,2,3-triazolylidene (tzNHCs) moieties, from Ag(I) to Ru(II) provided access to well-defined cationic [RuII(CO)(H)(L)(PPh3)]+ complexes 2 and 5. Spectroscopic investigations confirm that, in both complexes, the tridentate ligand binds in a rare facial mode to the metal center. The complexes, that exhibit ligand-based reversible deprotonation/dearomatization reactivity, are active in catalytic ester hydrogenation in the presence of KOtBu (≥20 mol%) as an exogenous base. The beneficial effect of the base on catalytic activity relates to transesterification of substrates to the corresponding tert-butyl ester derivatives, which are hydrogenated considerably faster than methyl esters. The mechanistic findings in this work confirm that this transformation is very complex, with this transesterification, metal-ligand cooperative reactivity, base strength and possibly product inhibition all playing a role. Furthermore, relevant Ru(CNC)(hydride) species have been observed by NMR spectroscopy under near-catalytic conditions.The synthesis of novel bis(triazolyl)pyridine ligands and their coordination chemistry toward Ru(II) are discussed, and these complexes are shown to be active catalysts for the hydrogenation of carboxylic esters.Download high-res image (169KB)Download full-size image
Co-reporter:Ruben M. Drost;Daniël L. J. Broere;Jorin Hoogenboom;Simone N. de Baan;Martin Lutz;B. de Bruin;C. J. Elsevier
European Journal of Inorganic Chemistry 2015 Volume 2015( Issue 6) pp:982-996
Publication Date(Web):
DOI:10.1002/ejic.201403104
Abstract
We have studied the use of amino acid histidine as a precursor for N-heterocyclic carbene (NHC) ligands. This natural amino acid possesses an imidazole substituent, which makes it an interesting NHC precursor that contains both an acid and an amino functionality. These functionalities may be used for further tuning of NHC complexes. We have developed routes for the synthesis of symmetric and dissymmetric alkyl, benzyl, and aryl-substituted histidinium salts. Subsequently, the corresponding Ag and Pd histidylidenes were synthesized and the palladium complexes were tested in the Z-selective transfer semihydrogenation of alkynes. Histidylidene palladium complexes that contain additional donor functionalities were found to display good selectivities. The best catalytic results were obtained with a Pd-histidylidene complex that contains two picolyl functional groups.
Co-reporter:Soraya N. Sluijter;Lukas J. Jongkind ;Cornelis J. Elsevier
European Journal of Inorganic Chemistry 2015 Volume 2015( Issue 18) pp:2948-2955
Publication Date(Web):
DOI:10.1002/ejic.201500331
Abstract
Despite the prolific use of (di-)NHC complexes in homogeneous catalysis, there are relatively few reports on their successful application in asymmetric transformations. In this work the atropisomeric binaphthyl backbone was combined with readily obtainable 1,2,3-triazolylidenes to develop a strongly electron-donating C2-symmetric ligand. The ligand was efficiently synthesized in a three-step procedure in an overall yield of 91 % starting from commercially available materials. Strategies for the synthesis of the corresponding di-NHC silver(I), palladium(II), rhodium(I), and iridium(I) complexes have been developed. The rhodium(I) complex was employed in the catalytic asymmetric hydrosilylation of ketones, providing good conversions at catalyst loadings as low as 0.2 mol-% and giving chiral inductions of up to 51 % ee.
Co-reporter:Dr. Ruben M. Drost;Vera Rosar;Silvia Dalla Marta;Dr. Martin Lutz;Dr. Nicola Demitri;Dr. Barbara Milani;Dr. Bas de Bruin;Dr. Cornelis J. Elsevier
ChemCatChem 2015 Volume 7( Issue 14) pp:2095-2107
Publication Date(Web):
DOI:10.1002/cctc.201500200
Abstract
A protocol was developed to distinguish between well-defined molecular and nanoparticle-based catalysts for the Pd-catalyzed semihydrogenation reaction of alkynes to Z-alkenes. The protocol applies quantitative partial poisoning and dynamic light scattering methods, which allow the institution of additional validation experiments. For the quantitative partial poisoning method, tetramethylthiourea (TMTU) was developed as an alternative for the standard poison ligand CS2, and was found to be superior in its applicability. The protocol and the TMTU poison ligand were validated using the well-described [PdII(phenanthroline)]-catalyzed copolymerization of styrene and CO, confirming that this system is clearly operating as a well-defined molecular catalyst. The protocol was subsequently applied to three catalyst systems used for the semihydrogenation of alkynes. The first was proposed to be a molecular [Pd0(IMes)] catalyst that uses molecular hydrogen, but the data gathered for this system, following the new protocol, clearly showed that nanoparticles (NPs) are catalytically active. The second catalyst system studied was an N-heterocyclic carbene (NHC) Pd system for transfer semihydrogenation using formic acid as the hydrogen source, which was proposed to operate through an in situ generated molecular [Pd0(IMes)] catalyst in earlier studies. The investigations showed that only a small fraction of the Pd added becomes active in the catalytic reaction and that NPs are formed. However, despite these findings, a clear distinction between catalytic activity of NPs versus a molecular catalyst could not be made. The third investigated system is based on a [PdII(IMes)(η3-allyl)Cl] precatalyst with additive ligands. The combined data gathered for this system are multi-interpretable, but suggest that a partially deactivated molecular catalyst dominates in this reaction.
Co-reporter:Ties J. Korstanje;Jarl Ivar van der Vlugt;Cornelis J. Elsevier;Bas de Bruin
Science 2015 Volume 350(Issue 6258) pp:298-302
Publication Date(Web):16 Oct 2015
DOI:10.1126/science.aaa8938
A direct route from acids to alcohols
Making alcohols via hydrogen addition to C=O bonds is among the most widely applied reactions in chemistry. The transformation has also garnered renewed interest for generating commodity chemicals from biomass. Korstanje et al. now show that a cobalt compound can catalyze hydrogenation of the C=O bonds in carboxylic acids. These constitute a particularly challenging substrate class, given the propensity of many other catalysts to degrade under acidic conditions. The cobalt catalyst tolerates a versatile substrate range, and the Earth abundance of the metal bodes well for long-term utility.
Science, this issue p. 298
Co-reporter:Ruben M. Drost, Tessel Bouwens, Nicolaas P. van Leest, Bas de Bruin, and Cornelis J. Elsevier
ACS Catalysis 2014 Volume 4(Issue 5) pp:1349
Publication Date(Web):March 20, 2014
DOI:10.1021/cs4011502
A convenient and easy-to-use protocol for the Z-selective transfer semihydrogenation of alkynes was developed, using ammonium formate as the hydrogen source and the easily prepared and commercially available, highly stable complex PdCl(η3-C3H5)(IMes) (1) as the (pre)catalyst. Combined with triphenyl posphine as an additional ligand, this system provides a robust catalytic synthetic method that shows little to no over-reduction or isomerization after full substrate conversion. The system allows the direct use of solvents and reagents, as received from the supplier without drying or purification, thus providing a practical method for semihydrogenation of a broad range of alkynes. The mechanism behind these high and enhanced selectivities was determined through a set of kinetic experiments.Keywords: alkyne reduction; N-heterocyclic carbene; NHC; palladium; transfer-hydrogenation
Co-reporter:Eveline Jansen, Martin Lutz, Bas de Bruin, and Cornelis J. Elsevier
Organometallics 2014 Volume 33(Issue 11) pp:2853-2861
Publication Date(Web):May 22, 2014
DOI:10.1021/om5003599
The development of a novel set of complexes bearing an NHC-amine ligand (CNHC-NH2) is described. M(cod) complexes (M = Ir, Rh) and a Ru complex have been synthesized in which three different coordination modes of the ligand were established: monodentate, neutral bidentate, and anionic bidentate. The anionic bidentate coordination mode of the anionic CNHC-NH– ligand arises from deprotonation of the amine moiety of the neutral CNHC-NH2 ligand. Ligand deprotonation proved to be reversible for the Rh and Ir complexes, as was shown by subsequent treatment of the complexes with base and acid. The structural parameters of these differently coordinated ligands were examined, and it was shown that the conjugation of the aniline ring plays a major role in determining the ligand properties. Structural parameters derived from DFT calculations confirm delocalization of the anionic charge over the ligand framework, as is clear from a comparison of the (hypothetical) neutral bidentate complexes [M(cod)(κ2C,N-{CNHC-NH2})]+ with those of the (synthesized) monoanionic complexes [M(cod)(κ2C,N-{CNHC-NH})] (M = Rh, Ir). A similar trend in the structure and bond lengths of the aniline rings was found in the solid-state structure of the novel dimeric complex [(Ru(κ2C,N-{CNHC-NH})(κ2C,N-{CNHC-NH2})Cl)2(μ-Cl)](PF6). The octahedral d5 ruthenium(III) centers in this complex both contain a neutral bidentate CNHC-NH2 ligand as well as an anionic bidentate CNHC-NH– ligand. Quite remarkably, the complex is diamagnetic, arising from antiferromagnetic coupling of the two low-spin ruthenium(III) centers over the chloride linker. DFT calculations indeed confirm that the open-shell singlet electronic structure is most stable.
Co-reporter:Soraya N. Sluijter and Cornelis J. Elsevier
Organometallics 2014 Volume 33(Issue 22) pp:6389-6397
Publication Date(Web):October 27, 2014
DOI:10.1021/om5007038
1,2,3-Triazol-5-ylidenes (tzNHC) have become a popular class of NHC ligands in homogeneous catalysis. Herein, we introduce chelate monovalent Rh- and Ir(cod) complexes bearing bidentate ligands that combine this tzNHC and an Arduengo-type NHC motif. The reactivity of these complexes with H2 and CO gas has been investigated, leading to an interesting octahedral [Ir(tzNHC-CH2-NHC)(CO)2(H)2]OTf complex and [M(tzNHC-CH2-NHC)(CO)2]OTf complexes. The carbonyl stretching frequencies of the latter indicate that the ligand has stronger electron-donating properties than classic di-NHC ligands. The square planar rhodium and iridium NHC-tzNHC complexes have been applied in transfer hydrogenation employing isopropyl alcohol as the hydrogen donor, in which they show moderate activity (Ir > Rh) toward a range of ketones as well as for an aldehyde, an imine, and a diene. The new dicarbene complexes proved to be more active for this reaction than the analogues in which the triazolyl moiety coordinates through a nitrogen donor.
Co-reporter:Soraya N. Sluijter, Stefan Warsink, Martin Lutz and Cornelis J. Elsevier
Dalton Transactions 2013 vol. 42(Issue 20) pp:7365-7372
Publication Date(Web):16 Jan 2013
DOI:10.1039/C3DT32835J
A transmetallation route, using silver(I) precursors, to several zero- and di-valent palladium complexes with chelating bis(N-heterocyclic carbene) ligands bearing various N-substituents has been established. The resulting complexes have been characterized by NMR and mass spectroscopy. In addition, the structure of a representative compound, [Pd0(bis-(Mes)NHC)(η2-ma)] (3a), was confirmed by X-ray crystal structure determination. In contrast to the transfer semihydrogenation, in which only low activity was observed, complex 3a showed activity (TOF = 49 molsub molcat−1 h−1) and selectivity comparable to its monodentate counterparts in the semihydrogenation of 1-phenyl-1-propyne with molecular hydrogen.
Co-reporter:Eveline Jansen;Linda S. Jongbloed;Dr. Dorette S. Tromp;Dr. Martin Lutz;Dr. Bas deBruin;Dr. Cornelis J. Elsevier
ChemSusChem 2013 Volume 6( Issue 9) pp:1737-1744
Publication Date(Web):
DOI:10.1002/cssc.201300363
Abstract
We herein report on the application and structural investigation of a new set of complexes that contain bidentate N-heterocyclic carbenes (NHCs) and primary amine moieties of the type [M(arene)Cl(L)] [M=Ru, Ir, or Rh; arene=p-cymene or pentamethylcyclopentadienyl; L=1-(2-aminophenyl)-3-(n-alkyl)imidazol-2-ylidine]. These complexes were tested and compared in the hydrogenation of acetophenone with hydrogen. Structural variations in the chelate ring size of the heteroditopic ligand revealed that smaller chelate ring sizes in combination with ring conjugation in the ligand are beneficial for the activity of this type of catalyst, favoring an inner-sphere coordination pathway. Additionally, increasing the steric bulk of the alkyl substituent on the NHC aided the reaction, showing almost no induction period and formation of a more active catalyst for the n-butyl complex relative to complexes with smaller Me and Et substituents. As is common in hydrogenation reactions, the activity of the complexes decreases in the order Ru>Ir>Rh. The application of [Ru(p-cym)Cl(L)]PF6, which outperforms its reported analogues, has been successfully extended to the hydrogenation of more challenging biomass-inspired substrates.
Co-reporter:Peter Hauwert, Jay J. Dunsford, Dorette S. Tromp, Jan J. Weigand, Martin Lutz, Kingsley J. Cavell, and Cornelis J. Elsevier
Organometallics 2013 Volume 32(Issue 1) pp:131-140
Publication Date(Web):December 19, 2012
DOI:10.1021/om300930w
In search of more active catalysts for the transfer hydrogenation of alkynes, a series of [Pd(NHC)(MA)1,2] (8–14) and [Pd(NHC)(dvtms)] complexes (1–7), in which the NHC ancillary ligands are expanded-ring N-heterocyclic carbenes (erNHC’s), have been prepared. These very bulky, strong σ-donor ligands impart a highly constrained geometry on the complexes and in some cases enable the isolation of coordinatively and electronically unsaturated complexes (10 and 14). Their strong σ-donor character is reflected in a decrease in IR stretching frequency for the C═O bond of the maleic anhydride ligands (8–14) in comparison to their five-membered counterparts. Significantly enhanced catalytic activity in the transfer hydrogenation of 1-phenyl-1-propyne is observed using [Pd(erNHC)(dvtms)] complexes (1–7) as precatalysts. The catalysts show high initial selectivity toward (Z)-alkene. However, double-bond isomerization and over-reduction to the corresponding alkane occur when all the alkyne substrate is consumed; this feature reflects the very high efficiency of these catalysts in the transfer hydrogenation of alkynes as well as alkenes.
Co-reporter:Stefan Warsink;Suzanne Bosman;Jan J. Weig;Cornelis J. Elsevier
Applied Organometallic Chemistry 2011 Volume 25( Issue 4) pp:276-282
Publication Date(Web):
DOI:10.1002/aoc.1754
Abstract
The synthesis of an air-stable series of Pd0 complexes with unsymmetric bidentate N-pyridine N-heterocyclic carbene ligands has been described. The carbenes were generated by synthesis of the silver(I) complexes from the imidazolium salts, followed by transmetallation of the C-N ligands to obtain the target electron-rich zerovalent palladium compounds. The bidentate coordination behaviour of the ligands was confirmed by 1H, 13C NMR and X-ray spectroscopy. The complexes are active precatalysts for the selective transfer semihydrogenation of alkynes to Z-alkenes, with selectivities up to 99%. Copyright © 2011 John Wiley & Sons, Ltd.
Co-reporter:Peter Hauwert ; Romilda Boerleider ; Stefan Warsink ; Jan J. Weigand ;Cornelis J. Elsevier
Journal of the American Chemical Society 2010 Volume 132(Issue 47) pp:16900-16910
Publication Date(Web):November 8, 2010
DOI:10.1021/ja1062407
The transfer semihydrogenation of alkynes to (Z)-alkenes shows excellent chemo- and stereoselectivity when using a zerovalent palladium(NHC)(maleic anhydride)-complex as precatalyst and triethylammonium formate as hydrogen donor. Studies on the kinetics under reaction conditions showed a broken positive order in substrate and first order in catalyst and hydrogen donor. Deuterium-labeling studies on the hydrogen donor showed that both hydrogens of formic acid display a primary kinetic isotope effect, indicating that proton and hydride transfers are separate rate-determining steps. By monitoring the reaction with NMR, we observed the presence of a coordinated formate anion and found that part of the maleic anhydride remains coordinated during the reaction. From these observations, we propose a mechanism in which hydrogen transfer from coordinated formate anion to zerovalent palladium(NHC)(MA)(alkyne)-complex is followed by migratory insertion of hydride, after which the product alkene is liberated by proton transfer from the triethylammonium cation. The explanation for the high selectivity observed lies in the competition between strongly coordinating solvent and alkyne for a Pd(alkene)-intermediate.
Co-reporter:Stefan Warsink, Ruben M. Drost, Martin Lutz, Anthony L. Spek and Cornelis J. Elsevier
Organometallics 2010 Volume 29(Issue 14) pp:3109-3116
Publication Date(Web):June 22, 2010
DOI:10.1021/om100435x
New heterobidentate N-heterocyclic carbene-triazolyl ligands and several of their palladium(II) complexes have been synthesized in a modular fashion using click chemistry. These complexes are the first examples where triazolyl-substituted NHCs exhibit bidentate behavior, which was confirmed by NMR and X-ray diffraction studies. The synthesis of the complexes could be achieved in relatively few steps by introducing the diversity at a late stage in the synthesis. The complexes are active precatalysts in the transfer semihydrogenation of alkynes to Z-alkenes, with activity and selectivity depending on the triazolyl substituent and the NHC nitrogen substituent. Selectivities as high as 99% were observed.
Co-reporter:Stefan Warsink, I-Hsin Chang, Jan J. Weigand, Peter Hauwert, Jwu-Ting Chen, and Cornelis J. Elsevier
Organometallics 2010 Volume 29(Issue 20) pp:4555-4561
Publication Date(Web):October 1, 2010
DOI:10.1021/om100670u
Several (N-2-pyrimidyl-NHC)-palladium(0) complexes have been synthesized in very high yields and purities by transmetalation from the related silver(I) complexes with Pd0(tBuDAB)(ma). The coordination behavior of the heteroditopic ligands was analyzed, and the structure of their complexes was confirmed by NMR and X-ray diffraction studies. The complexes are active catalysts for the transfer hydrogenation of alkynes to Z-alkenes, with activity and selectivity depending on the pyrimidine substituents and the NHC nitrogen substituent. Selectivities toward the Z-alkene as high as 97% were observed.
Co-reporter:Stefan Warsink, Sandra Y. de Boer, Lianne M. Jongens, Ching-Feng Fu, Shiuh-Tzung Liu, Jwu-Ting Chen, Martin Lutz, Anthony L. Spek and Cornelis J. Elsevier
Dalton Transactions 2009 (Issue 35) pp:7080-7086
Publication Date(Web):21 Jul 2009
DOI:10.1039/B906817A
A number of palladium(II) complexes with a heteroditopic NHC-amine ligand and their precursor silver(I) carbene complexes have been efficiently prepared and their structural features have been investigated. The heteroditopic coordination of this ligand class was unequivocally shown by NMR-spectroscopy and X-ray crystallographic analysis. The neutral and cationic cis-methyl-palladium(NHC) complexes are not prone to reductive elimination, which is normally a major degenerative pathway for this type of complex. In contrast, under carbon monoxide atmosphere rapid reductive elimination of the acyl-imidazolium salt was observed.
Co-reporter:Michael R. Eberhard;Bart van Vliet;Laura Durán Páchon;Gadi Rothenberg;Graham Eastham;Huub Kooijman;Anthony L. Spek;Cornelis J. Elsevier
European Journal of Inorganic Chemistry 2009 Volume 2009( Issue 10) pp:1313-1316
Publication Date(Web):
DOI:10.1002/ejic.200801067
Abstract
We present a straightforward protocol for making (phosphanyl-carbene)PdII complexes. These complexes have bidentate ligands containing an acyclic diamino- or aminooxy-carbene and a phosphane. The synthesis gives good yields (typically 70–90 %) for a variety of complexes (22 compounds). Moreover, it does not require the synthesis of imidazolium salts nor the a priori generation of free carbenes. Three of the new complexes were tested as catalysts for Sonogashira and Hay coupling reactions, with good yields and selectivities. (© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2009)
Co-reporter:Soraya N. Sluijter, Stefan Warsink, Martin Lutz and Cornelis J. Elsevier
Dalton Transactions 2013 - vol. 42(Issue 20) pp:NaN7372-7372
Publication Date(Web):2013/01/16
DOI:10.1039/C3DT32835J
A transmetallation route, using silver(I) precursors, to several zero- and di-valent palladium complexes with chelating bis(N-heterocyclic carbene) ligands bearing various N-substituents has been established. The resulting complexes have been characterized by NMR and mass spectroscopy. In addition, the structure of a representative compound, [Pd0(bis-(Mes)NHC)(η2-ma)] (3a), was confirmed by X-ray crystal structure determination. In contrast to the transfer semihydrogenation, in which only low activity was observed, complex 3a showed activity (TOF = 49 molsub molcat−1 h−1) and selectivity comparable to its monodentate counterparts in the semihydrogenation of 1-phenyl-1-propyne with molecular hydrogen.
Co-reporter:Stefan Warsink, Sandra Y. de Boer, Lianne M. Jongens, Ching-Feng Fu, Shiuh-Tzung Liu, Jwu-Ting Chen, Martin Lutz, Anthony L. Spek and Cornelis J. Elsevier
Dalton Transactions 2009(Issue 35) pp:NaN7086-7086
Publication Date(Web):2009/07/21
DOI:10.1039/B906817A
A number of palladium(II) complexes with a heteroditopic NHC-amine ligand and their precursor silver(I) carbene complexes have been efficiently prepared and their structural features have been investigated. The heteroditopic coordination of this ligand class was unequivocally shown by NMR-spectroscopy and X-ray crystallographic analysis. The neutral and cationic cis-methyl-palladium(NHC) complexes are not prone to reductive elimination, which is normally a major degenerative pathway for this type of complex. In contrast, under carbon monoxide atmosphere rapid reductive elimination of the acyl-imidazolium salt was observed.