Asymmetric formylation of aromatic compounds is virtually unexplored. We report the synthesis and evaluation of a library including 20 new chiral formamides in the kinetic resolution of 7,8-dipropyltetrathia[7]helicene, affording the corresponding formyl- or diformylhelicenes in up to 73 % ee, making enantiopure compounds available by recrystallisation. With the N,N-disubstituted formamides used in this study, the best enantioselectivity has been achieved with R1=iPr, R2=Me, R3=H, R4=1-naphthyl or its 1-pyrenyl equivalent.
The effect of organolithium reagent (RLi: R=nBu, iPr, sBu, tBu), solvent (diethyl ether, diethyl ether/THF and MTBE), and stoichiometry on the (−)-sparteine-mediated silylation of 7,8-dipropyltetrathia[7]helicene shows that, unusually, substantially more than 0.5 equivalent of RLi (R=iPr, sBu, tBu) and a large excess of (−)-sparteine (R=nBu, sBu) is often needed to achieve substantial conversions and good ee values. With nBuLi, however, just one equivalent of the organolithium reagent is sufficient to obtain high conversions. Our best results were obtained using the convenient tBuLi/(−)-sparteine adduct with which the need for a high (−)-sparteine/RLi ratio can be avoided. Single- and double-kinetic resolution (KR) procedures give enantiopure samples of 2-trimethylsilyl- and 2,13-di(trimethylsilyl)-7,8-dipropyltetrathia[7]helicene and two-step double-KR combining (−)-sparteine/sBuLi and chiral formamides affords the synthetically valuable 2-formyl-7,8-dipropyltetrathia[7]helicene. This is the first use of (−)-sparteine for the enantioselective lithiation of helicenes and the first report of tBuLi outperforming sBuLi in a (−)-sparteine-mediated procedure.
Disubstituted (cyclohexadienyl)iron(1+) complex 2b is prepared by an improved route that starts from 1,2-dimethoxycyclohexa-1,4-diene 3. In five steps, the synthesis of 2b is achieved by complexation with Fe(CO)5, hydride abstraction, hydrolysis, addition of EtO2CCH2ZrBr, and reaction with HBF4. In the presence of dimethyl sulfide, the reaction of 2b with 2-[CH2N(CH2CH=CH2)2]-functionalized diarylcuprate reagent 7 gave the 5α-arylcyclohexadiene complex 1b in 88 % yield. A DFT study compared diarylzinc and diarylcuprate reagents containing chelating CH2NMe2 substituents.
Aryllithium reagents react with tricarbonyl(η5-1,4-dimethoxycyclohexadienyl)iron hexafluorophosphate to give η4 products, which are converted into a series of (1-arylcyclohexadienyl)iron(1+) complexes with H, CHO, CH2OAc, and CH(OAc)2 on the arene, ortho to the point of attachment to the dienyl ligand. The structures of these complexes have been compared by X-ray crystallography. Correlations between spectroscopic and structural data indicate that the crystallographically defined conformations are also significant in the interpretation of the pathways of nucleophile addition in solution. Reactions with nucleophiles are influenced by the conformational properties of the 1-aryl substituent, and examples of ω addition and a new intramolecular ipso addition relative to the arene have been characterised. Intermolecular ipso addition has been used to synthesise the alkaloid mesembrine in which 3,4-dimethoxy electron-donating substituents flatten the arylcyclohexadienyl portion and facilitate the ipso pathway. Electron-withdrawing formyl substituents cause the arene to adopt a more perpendicular conformation, and a through-space interaction between CHO and the exo face of the cationic dienyliron complex activates the aldehyde for reaction with (PhC≡C)2CuLi, generating an alkoxide that forms a spirocyclic ether by intramolecular reaction with the aryl-substituted terminus of the dienyl ligand. Full spectroscopic assignments of the (1-arylcyclohexadienyl)iron(1+) complexes are reported and correlated with structural data from crystallography to show that the complexes fall into two groups in terms of the relationship between the dihedral angle of the arene and the dienyl systems and the weighted means for ν(CO) in their infrared spectra. The relationship between this structural data, the distribution of positive charge on the dienyliron complex, and the regioselectivity of nucleophile addition explains how the correct choice of substitution patterns influences the selectivity between ipso and ω reactivity in 1,1-iterative strategies that use (1-arylcyclohexadienyl)iron(1+) complexes as key building blocks in synthetic routes to the galanthamine and crinine subclasses of the Amaryllidaceae alkaloids.
FTIR spectra of tricarbonyl(η6-benzo-15-crown-5)chromium(0) (1) in the presence of lithium, sodium and potassium perchlorate salts in methanol show different responses in the Cr–CO vibrational region of the spectrum. Data from the symmetric (νsym) and antisymmetric (νasym) Cr–CO vibrational stretching modes have been analysed by principal component analysis (PCA) to generate a factor score plot that provides a visual representation of these differential responses. X-ray crystallographic data for the sodium perchlorate complex 1·Na+ and dimensions from DFT-derived structures of 1·Li+, 1·Na+ and 1·K+ indicate that binding M+ in the crown causes electron density and structural changes in the [O(4)–C(9)–C(4)–O(8)]Cr–C(1)=O(1) sections of 1, which vary depending on the nature of the cation. This suggests a mode of action in which Li+ associates with a more compact O(4)–C(9)–C(4)–O(8), while Na+ and K+ differ crucially in the extent of σ and π contributions to their effect on νsym and νasym. A comparison of the FTIR data from 1, tricarbonyl(η6-1-phenyl-1-aza-15-crown-5)chromium(0) (2) and tricarbonyl(η6-2-phenyl-15-crown-5)chromium(0) (3) with a wider range of cations (NH4+, Li+, Na+, K+, Rb+, Cs+, Mg2+, Ba2+) and anions (AcO–, BPh4–, Br–, C1O4–, I–, SCN–), showed that 1 and 3 both responded significantly to the different metal cations, but 2 did not. The relative cation differentiation of 1, 2 and 3 was measured using the parameter ΔR(cation), and ratios of ΔN(cation) values [calculated from ΔR(cation)] distinguished different effects in the FTIR spectra of 1 and 3 for different pairs of cations.
Aryllithium reagents generated from protected 6-bromoguaiacol and 2-bromo-4,5-dimethoxybenzyl alcohol derivatives were used to prepare ortho-substituted (1-arylcyclohexadienyl)iron(1+) electrophiles. These were treated with Na+[Me3SiCH2CH2O2CCHCN]– to build aryl-substituted quaternary centres in new examples of 1,1 iterative {[η4] [η5]+ [η4] [η5]+ [η4]} reaction sequences, which make use of the electrophilicity of the metal complex in two key carbon–carbon bond-formation steps. MOM protection of the guaiacol was better than SEM for access to the lycoramine skeleton, and TBDPS was best for maritidine. Decomplexation, hydrolysis, and cyclisation completed formal total syntheses of the Amaryllidaceae alkaloids (±)-lycoramine and (±)-marididine, establishing the compatibility of the organoiron method with the presence of ortho substituents on the aryl group, and nucleophile addition ipso to the substituted arene.
A crystallographic investigation comparing five 1-aryl-substituted tricarbonyl[(1–5-η)-cyclohexadienyl]iron(1+) salts demonstrates that introducing additional electron density on the aromatic ring increases π overlap between the arene and the cyclohexadienyl ligand, thus flattening the structures sufficiently to make available a conformation in which nucleophiles can approach the site of substitution, despite the steric blockade of o-benzyl substituents. (© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2008)
A series of aryl-substituted cyclohexadienyliron complexes have been prepared by a general procedure that determines regioselectivity by correctly positioning leaving groups in the precursor complexes. The aryl groups at 1-C or 2-C have been shown to be ω directing by the study of reactions with a representative range of nucleophiles, and these regioselectivity properties have been related to the spectroscopic properties of the cationic cyclohexadienyliron complexes. A high level of electron-donating substituents on the arene, or switching between the [Fe(CO)3] and [Fe(CO)2PPh3] series, reduces the minor ipso pathway, improving regiocontrol. Placing opposed directing groups in the arylcyclohexadienyliron complexes reverts reactivity to the ipso pathway with stabilised enolate nucleophiles, and when the additional directing group reinforces the effect of the aryl group, the ipso pathway is stopped.
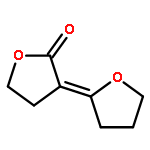
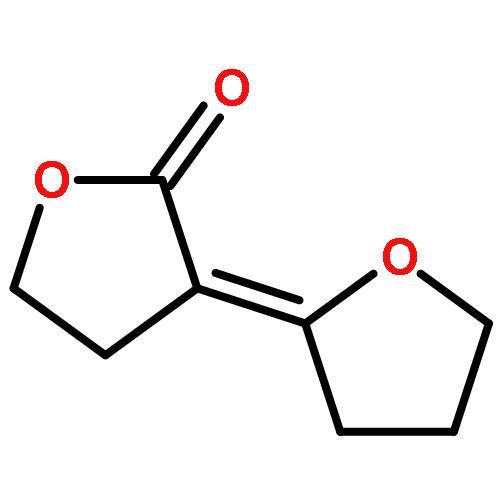
Potential “reagents” for the enantioselective reduction, and other biotransformations, of β-keto-esters result from the genetic engineering of Streptomyces coelicolor A3(2). For example, incubation of the N-acetylcysteamine thioester 1 with the recombinant strain CH999/pIJ5675 followed by treatment with MeOH/HCl gave the lactone 2 as essentially a single enantiomer.
Potentielle „Reagentien” für die enantioselektive Reduktion und andere Biotransformationen von β-Ketoestern entstehen bei der gentechnischen Veränderung von Streptomycescoelicolor A3(2). Beispielsweise lieferten die Inkubation des Thioesters 1 mit dem rekombinanten Stamm CH999/pIJ5675 und die anschließende Behandlung mit MeOH/HCl das Lacton 2 nahezu enantiomerenrein.
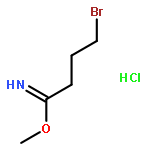
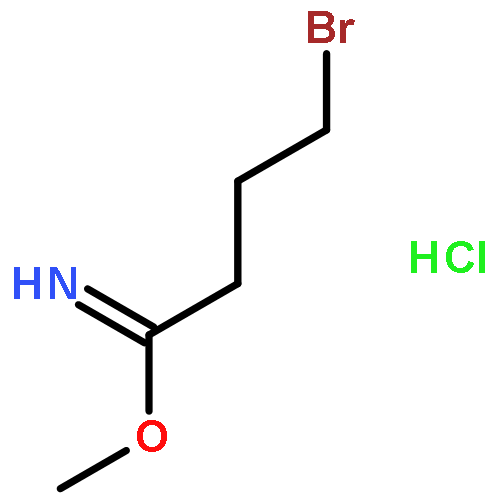


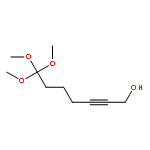
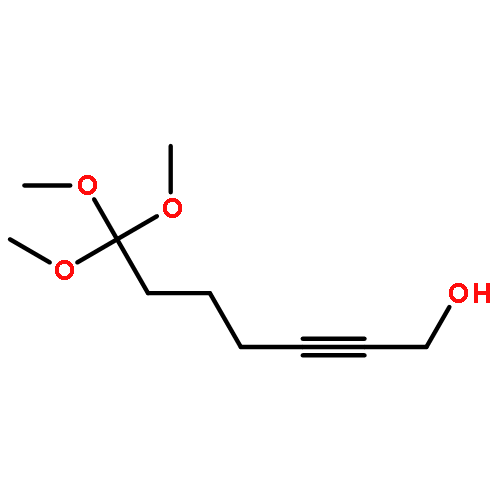
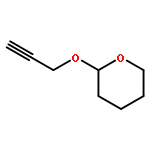
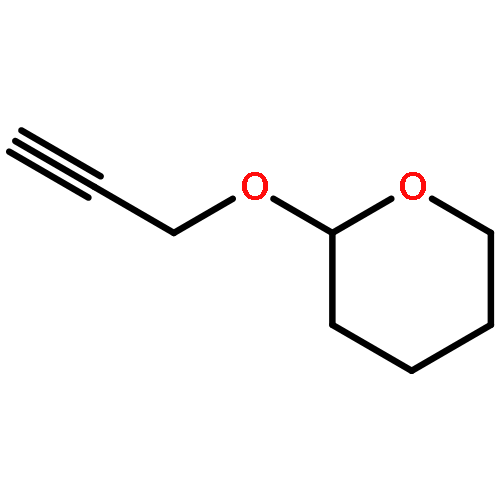
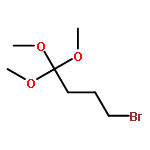
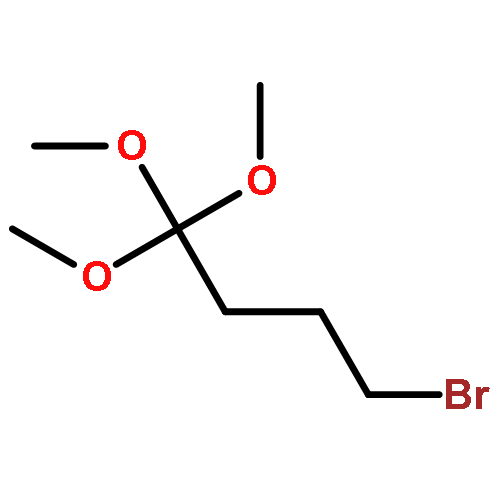
![5-Heptynoic acid, 7-[(tetrahydro-2H-pyran-2-yl)oxy]-, methyl ester](http://img.cochemist.com/ccimg/50800/50781-90-3.png)
![5-Heptynoic acid, 7-[(tetrahydro-2H-pyran-2-yl)oxy]-, methyl ester](http://img.cochemist.com/ccimg/50800/50781-90-3_b.png)