Co-reporter:Anton J. Stasyuk, Piotr J. Cywiński, Daniel T. Gryko
Journal of Photochemistry and Photobiology C: Photochemistry Reviews 2016 Volume 28() pp:116-137
Publication Date(Web):September 2016
DOI:10.1016/j.jphotochemrev.2016.05.003
•ESIPT-capable compounds undergo aggregation-induced emission enhancement.•Incredible sensitivity of optical properties towards substituents effect.•Novel synthetic methodologies make it possible to broaden the scope of studied derivatives.The key observation made by Acuña revealed the unusual optical properties of previously unknown 2-(2′-hydroxyphenyl)imidazo[1,2-a]pyridines (HPIPs). Although structurally similar to 2′-(2′-hydroxyphenyl)benzoxazoles and 2-(2′-hydroxyphenyl)benzimidazoles, 2-(2′-hydroxyphenyl)imidazo[1,2-a]pyridines exhibit strikingly different optical properties, especially in the solid state. The luminescence color of HPIPs depends on the polymorphic form and can be tailored from green to red. The inability to assume the neutral keto tautomer is their other distinct characteristic, setting them apart from analogous azoles and their benzoanalogs. For these azoles and related luminescent compounds, the dual emission in polar solvents is usually observed, dominated by excited-state intramolecular proton transfer (ESIPT) fluorescence. However, for 2-(2′-hydroxyphenyl)imidazo[1,2-a]pyridines, clear ESIPT emission exists only in non-polar solvents and it is very weak, while in polar and especially protic solvents, strong emission from the Franck-Condon state is exclusively observed. The solvent-related effect is attributed to the tendency to form strong intramolecular hydrogen bonds in the solid state and in non-polar solvents, while hydrogen bond formation is much weaker in polar, and especially, in protic solvents. The present study focuses on HPIPs and demonstrates that ESIPT process is a promising mechanism for packing-directed luminescence control and offers a novel design concept for tunable organic luminescent solids. This review presents not only optical properties in various solutions, but also in the solid state, and it is supported by a synthetic overview, computational studies, and the description of π-expanded derivatives.
Co-reporter:Bartosz Bursa, Bolesław Barszcz, Waldemar Bednarski, Jan Paweł Lewtak, Dominik Koszelewski, Olena Vakuliuk, Daniel T. Gryko and Danuta Wróbel
Physical Chemistry Chemical Physics 2015 vol. 17(Issue 11) pp:7411-7423
Publication Date(Web):04 Feb 2015
DOI:10.1039/C4CP05648E
The investigation presented in this paper deals with new free-base corroles substituted with different peripheral groups. These aromatic macrocycles were efficiently synthesized by a [2+1] approach from dipyrromethanes. Moreover, the basic spectroscopic studies of the dyes in chloroform were conducted, and the UV-Vis absorption, fluorescence and ESR parameters were estimated. The experimental data were supported by quantum chemical calculations. The presence of monomeric dye structures is concentration independent (10−6–10−4 M), as expected for dyes in a solvent of low polarity, and rules out aggregate formation of corroles dissolved in chloroform. The excitation emission and fluorescence life-time values confirm the monomeric structure of the corroles. The spectra were compared with the time-dependent density functional theory (TD-DFT) results for the HOMO–LUMO states. The ESR examinations strongly show that for any type of studied fluorine corrole an unpaired electron is localized on the corrole macroring but not on the substituents both before and after light illumination. Laser illumination creates additional radicals, however with different effectiveness depending on the sample.
Co-reporter:Kornelia Lewandowska, Bolesław Barszcz, Andrzej Graja, Bartosz Bursa, Andrzej Biadasz, Danuta Wróbel, Waldemar Bednarski, Stefan Waplak, Marek Grzybowski, Daniel T. Gryko
Synthetic Metals 2013 Volume 166() pp:70-76
Publication Date(Web):15 February 2013
DOI:10.1016/j.synthmet.2013.01.014
The bichromophoric systems comprising of corrole and fullerene units and representing one of the rare cases of elaborate structures based on corrole have been studied with the use of photophysical methods. The dyad displays spectroscopic properties which are the superposition of the component spectra, indicating a very weak electronic coupling. Excitation of the corrole unit leads to charge redistribution. The results of UV–vis absorption and fluorescence and also fluorescence kinetics suggest the presence of charge separation. It was shown that light absorption of the corrole–fullerene dyad is better fitted to the sunlight spectrum than porphyrin. Electron paramagnetic resonance investigations suggest partial spin density redistribution at low temperature between corrole and fullerene in the dyad. Quantum chemical calculations support experimental results.Graphical abstractHighlights► Extended spectroscopic and quantum chemical investigations of a new corrole–fullerene dyad. ► Photoinduced electron transfer and charge localization in the corrole–fullerene dyad. ► First studies of UV–vis absorption, fluorescence, fluorescence kinetics and EPR spectra. ► Corrole–fullerene dyad perfectly fitted to the sunlight and suitable for light conversion.
Co-reporter:Olena Vakuliuk;Beata Koszarna;Daniel T. Gryko
Advanced Synthesis & Catalysis 2011 Volume 353( Issue 6) pp:925-930
Publication Date(Web):
DOI:10.1002/adsc.201000723
Abstract
It appears that transition metal catalysts are not necessary to perform the direct arylation of electron-rich heterocycles with aryl iodides and bromides. Lithium tert-butoxide in DMF promotes this reaction for a variety of N-alkyl- and N-arylpyrroles as well as for benzofuran and some other electron-rich aromatic compounds and provides the desired products in moderate to high yields. In contrast to all previous reports on the Pd-catalyzed direct arylation of indolizine, the reaction mediated by lithium tert-butoxide proceeds selectively at position 5.
Co-reporter:Olena Vakuliuk ;Daniel T. Gryko
European Journal of Organic Chemistry 2011 Volume 2011( Issue 15) pp:2854-2859
Publication Date(Web):
DOI:10.1002/ejoc.201100004
Abstract
An efficient methodology for the direct arylation of pyrrole derivatives has been developed. The reaction proceeds smoothly with a wide range of structurally diverse aryliodides. Derivatives bearing an N,N-dimethylamino group at the 1-position and an aryl substituent at the 2-position were prepared for the first time. This protocol is more environmentally friendly than those previously reported because it is free of transition metals and utilizes ionic liquids rather than volatile organic solvents.
Co-reporter:Ayfer Kalkan Burat, Atıf Koca, Jan P. Lewtak, Daniel T. Gryko
Synthetic Metals 2011 Volume 161(15–16) pp:1537-1545
Publication Date(Web):August 2011
DOI:10.1016/j.synthmet.2011.05.010
The synthesis of novel, unsymmetrical, octasubstituted metal-free and metallophthalocyanines (zinc, cobalt) bearing one chlorine, one morpholine moieties and six 4-tert-butylphenoxy substituents was achieved by a statistical condensation reaction of two corresponding phthalonitriles. The new compounds have been characterized by using elemental analyses, UV–vis, IR, 1H NMR and mass spectroscopic data. Voltammetric and spectroelectrochemical studies are in harmony with the reported metallophthalocyanine complexes, which support the proposed structure of the complexes.
Co-reporter:Agnieszka Nowak-Król, Beata Koszarna, Su Yeon Yoo, Jan Chromiński, Marek K. Wȩcławski, Chang-Hee Lee, and Daniel T. Gryko
The Journal of Organic Chemistry 2011 Volume 76(Issue 8) pp:2627-2634
Publication Date(Web):March 16, 2011
DOI:10.1021/jo1025578
Efficient and convenient conditions for the preparation of trans-A2B2-porphyrins bearing two phenylethynyl moieties directly from phenylpropargyl aldehydes and dipyrromethanes of diversified lipophilicity and reactivity have been developed. This new procedure allows the preparation of a library of porphyrins of this architecture with a wide range of substituents. Thanks to the identification of the reagent solubility as one of the key factors influencing the yield of the porphyrinogens, we were able to improve yields to ca. 30%. The scope and limitations of two sets of conditions have been explored. The methodological advantage of this approach is its straightforward access to building blocks and the formation of the porphyrin core in the last step without the need for deprotection of the triple bond or bromination and consecutive coupling reaction, which often demands copper salts to proceed smoothly, especially with electron-deficient alkyne partners. Therefore, it prevents undesired copper porphyrin formation, as well as the need for utilizing expensive alkynes. A two-step method for the preparation of phenylpropargyl aldehydes has also been refined.
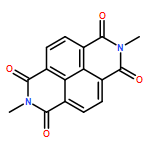
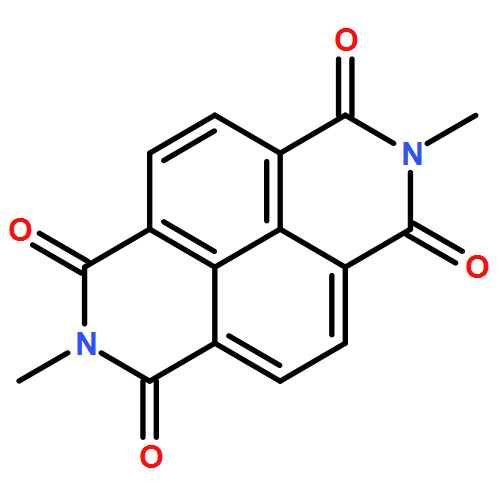
Co-reporter:Daniel T. Gryko, Maciej K. Rogacki, Jan Klajn, Michał Gałęzowski, Dorota K. Stȩpień and Michał K. Cyrański
Organic Letters 2010 Volume 12(Issue 9) pp:2020-2023
Publication Date(Web):April 13, 2010
DOI:10.1021/ol1005032
1,4,5,8-Naphthalene bisimides react as dipolarophiles with in situ formed azomethine ylides. Double 1,3-dipolar cycloaddition is followed by unique ring rearrangement and leads to the formation of two six-membered rings. The formation of hexacyclic products is rationalized based on DFT calculations.
Co-reporter:Lucia Flamigni, Dagmara Wyrostek, Roman Voloshchuk and Daniel T. Gryko
Physical Chemistry Chemical Physics 2010 vol. 12(Issue 2) pp:474-483
Publication Date(Web):12 Nov 2009
DOI:10.1039/B916525H
A dyad (C3–NI) based on corrole and naphthalene bisimide has been synthesized and its photoreactivity compared to that of the model component corrole (C3) and naphthalene bisimide (NI) in solvents of different polarity: toluene (TL) and dichloromethane (DCM). The major emitting species in NI solutions, in TL, is identified as a dimeric species (λ = 470 nm, τ = 2.3 ns) but traces of monomer can also be detected (λ ca. 390 nm, τ = 40 ps). In DCM the major emitting component is the monomer (λ = 383 nm, τ = 20 ps) but traces of different aggregates (λ = 540 and 570 nm, τ = 4.5 and 11 ns) are present. C3 has a fluorescence nearly unaffected by solvent polarity, with a maximum around 655 and a lifetime of 3.5 or 3.8 ns in DCM and TL, respectively. The dyad C3–NI does not appear to be affected by aggregation problems in any of the solvents. Excitation of the imide component in C3–NI (C3–1NI) results in an energy transfer to corrole (1C3–NI) with rate k = 2.0 × 1011 s−1 in both solvents. The latter state reacts further via a LUMO–LUMO electron transfer to the naphthalene bisimide yielding the charge separated state C3+–NI− (k = 1.8 × 109 s−1 in TL and k = 3.7 × 109 s−1 in DCM). The same type of reactivity is displayed by direct excitation of the corrole moiety in the dyad to 1C3–NI. C3+–NI− decays with a rate comparable to that of its formation in DCM (k = 4.0 × 109 s−1in DCM), precluding its accumulation, whereas it decays with a slower rate in TL (k = 7.1 × 108 s−1). The charge separated state recombines to the ground singlet state; recombination to the triplet state of corrole (excited state at the lowest energy) is in fact excluded on the basis of the experimentally determined triplet yields. The failure of the commonly used methods in the calculation of CS energy levels in apolar solvents is confirmed.
Co-reporter:Agnieszka Nowak-Król;Dorota Gryko Dr. ;DanielT. Gryko
Chemistry – An Asian Journal 2010 Volume 5( Issue 4) pp:904-909
Publication Date(Web):
DOI:10.1002/asia.200900693
Abstract
Meso-substituted A4-porphyrins bearing 3,4,5-trialkoxyphenyl substituents are efficiently synthesized and characterized. Porphyrins bearing twelve C10 and C11 alkyl chains turned out to be liquid at room temperature. The remaining porphyrins, bearing C8, C9, C12, and C18 alkyl chains, have low melting points and high solubility in nonpolar solvents. Their differential scanning calorimetry distinctly shows, in most cases, only one phase transition.
Zsyntetyzowano sześć mezo podstawionych porfiryn zawierających podstawniki 3,4,5-trialkoksyfenylowe. Porfiryny z łańcuchami alkilowymi zawierającymi dziesięć i jedenaście atomów węgla są cieczami w temperaturze pokojowej. Pozostałe cztery porfiryny wykazują bardzo dobrą rozpuszczalność w rozpuszczalnikach niepolarnych oraz niskie temperatury topnienia. Badania metodą skaningowej kalorymetrii różnicowej wykazały dla większości porfiryn tylko jedno przejście fazowe.
Co-reporter:Mariusz Tasior Dr.;DanielT. Gryko ;DominikaJ. Pielaci&x144;ska;Alberto Zanelli Dr.;Lucia Flamigni Dr.
Chemistry – An Asian Journal 2010 Volume 5( Issue 1) pp:130-140
Publication Date(Web):
DOI:10.1002/asia.200900345
Abstract
Four dyads comprised of corrole and coumarin units have been synthesised. Three coumarincarboxaldehydes were synthesized and transformed into the corresponding trans-A2B-corroles by reaction with 5-(pentafluorophenyl)dipyrromethane. It has been proven that this type of direct condensation can lead to the corresponding corroles in moderate yields. The reaction of hydroxybenzaldehydes with vinylphosphonium salts has been identified as the most general method for the preparation of formyl-coumarins with various patterns of substituents. The dyad consisting of ketobiscoumarin and corrole was synthesized by Sonogashira coupling. Spectroscopic and photophysical investigations revealed that there is an efficient energy transfer from the coumarin moiety to corrole in all four dyads. Energy transfer can be clearly ascribed to a dipole–dipole mechanism (Förster) for all dyads that contain luminescent coumarins and to an electron exchange mechanism (Dexter) for the dyad with the non-luminescent one. In the case of the dyad that bears coumarin with a hydroxy group at position 5, an electron-transfer was detected from corrole to coumarin. The latter process is possible because of the suitably low reduction potential of coumarins of this type.
Cztery układy dwuchromoforowe, składające się z jednostek korolowych i kumarynowych zsyntetyzowano poprzez kondensację formylokumaryn z 5-(pentafluorofenylo)dipirometanem. Udowodniono, że reakcja aldehydów hydroksybenzoesowych z solami winylofosfoniowymi jest najbardziej ogólną metodą syntezy formylokumaryn. Badania spektroskopowe i fotofizyczne wykazały istnienie transferu energii od układu kumaryny do pierścienia korolu. Transfer ten zachodzi według mechanizmu Förstera w przypadku gdy kumaryna wykazuje fluorescencję a według mechanizmu Dextera dla kumaryny niefluoryzującej. Dla układu dwuchromoforowego zawierającego kumarynę z grupą hydroksylową w pozycji 5 zachodzi również transfer elektronu w kierunku od korolu do kumaryny.
Co-reporter:Agnieszka Nowak-Król;Dorota Gryko Dr. ;DanielT. Gryko
Chemistry – An Asian Journal 2010 Volume 5( Issue 4) pp:
Publication Date(Web):
DOI:10.1002/asia.201090008
Co-reporter:Lucia Flamigni and Daniel T. Gryko
Chemical Society Reviews 2009 vol. 38(Issue 6) pp:1635-1646
Publication Date(Web):07 Apr 2009
DOI:10.1039/B805230C
The collection and conversion of light energy into chemical energy is based on the use of molecular structures of various complexity, where the absorbed light energy is first converted into an excited state able to undergo energy or electron transfer processes and finally it is stored in a charge separated state as chemical (electrochemical) potential. A bio-mimetic approach has seen tetrapyrroles number among the most common components of these arrays. This tutorial review reports on the use of relatively new tetrapyrroles, corroles, in this field. A brief presentation of the electrochemical and photophysical properties of the corrole units relevant to the application is presented together with a discussion on the photo- and thermal stability issues, followed by an overview of the activity and improvements in the preparation of photo-active molecular arrays containing corroles.
Co-reporter:Roman S. Czernuszewicz ; Vicky Mody ; Arkadiusz Czader ; Michał Gałęzowski ;Daniel T. Gryko
Journal of the American Chemical Society 2009 Volume 131(Issue 40) pp:14214-14215
Publication Date(Web):September 16, 2009
DOI:10.1021/ja906393r
Resonance Raman (RR) spectroscopy and density functional theory (DFT) calculations of oxochromium(IV,V) derivatives of 5,10,15-tris(pentafluorophenyl)corrole (tpfpc) are shown to provide useful information about the relative strength of the metal−oxo bond in high-valent CrIV versus CrV corroles. Isotope labeling of the terminal oxo group with 18O revealed that the CrV−oxo (perchromyl) stretch of (tpfpc)CrVO vibrates at a frequency of 986 cm−1 in carbon disulfide, consistent with a triply bonded CrV≡O unit. In contrast, an acetonitrile solution produced RR scattering that rapidly changed with the number of scans collected and eventually became dominated by an 18O-sensitive vibration at a significantly higher frequency of 1002 cm−1. On the basis of DFT calculations and the observed 18/16O isotopic shift, we assigned this new RR band at 1002 cm−1 in acetonitrile as the CrIV−oxo (chromyl) stretch of the autoreduced [(tpfpc)CrIVO]− product, which previously has been shown to form only during the course of the oxygen atom transfer (OAT) reaction with triphenylphosphine in acetonitrile or in the presence of a reducing chemical (cobaltocene) and electrochemical agents in other solvents. Consequently, RR observations indicate that the π-bonding character of the chromyl bond is actually increased relative to that of the perchromyl bond, which is of interest if the beneficial role of acetonitrile in OAT catalysis by high-valent oxochromium(IV,V) corroles is to be elucidated.
Co-reporter:Richard van Hameren, Johannes A. A. W. Elemans, Dagmara Wyrostek, Mariusz Tasior, Daniel T. Gryko, Alan E. Rowan and Roeland J. M. Nolte
Journal of Materials Chemistry A 2009 vol. 19(Issue 1) pp:66-69
Publication Date(Web):17 Oct 2008
DOI:10.1039/B812518J
The self-assembly of corrole trimers in solution and at solid–liquid interfaces is a process that depends on dewetting, hydrogen bonding and π–π interactions between the molecules forming columnar stacks, and lateral interactions between these stacks to generate higher order assemblies.
Co-reporter:Daniel T. Gryko, Olena Vakuliuk, Dorota Gryko and Beata Koszarna
The Journal of Organic Chemistry 2009 Volume 74(Issue 24) pp:9517-9520
Publication Date(Web):November 11, 2009
DOI:10.1021/jo902124c
A methodology that affords N-alkyl-2-arylpyrroles and N-aryl-2-arylpyrroles via direct coupling from aryl iodides has been developed. After examining various reaction parameters: solvent, ratio of reagents, catalyst, base and additives the optimal conditions for the condensation were identified. Two crucial factors, (a) anhydrous DMSO as solvent and (b) 5 M excess of pyrrole counterpart, were found to strongly influence the reaction outcome. The conditions identified (PdCl2(PPh3)2, AgOAc, anhyd DMSO, KF, 100 °C, 5 h) resulted in the formation of 2-arylpyrroles in 14−80% yield. Furthermore, the synthesis is compatible with electron-withdrawing and electron-donating groups on the aryl moiety.
Co-reporter:Beata Koszarna and Daniel T. Gryko
Chemical Communications 2007 (Issue 28) pp:2994-2996
Publication Date(Web):15 May 2007
DOI:10.1039/B703279J
The cascade reaction of sterically hindered dipyrromethanes and formaldehyde furnished meso(10)–meso(10′) linked corroles—a new type of porphyrinoid—compounds with interesting photophysical properties.
Co-reporter:Lucia Flamigni, Barbara Ventura, Mariusz Tasior, Daniel T. Gryko
Inorganica Chimica Acta 2007 Volume 360(Issue 3) pp:803-813
Publication Date(Web):15 February 2007
DOI:10.1016/j.ica.2006.03.021
The synthesis of a new stable corrole-porphyrin dyad, made up of a free-base corrole and a free-base porphyrin connected by an amide linker is presented and the characterization of the photoinduced processes taking place in the array is described. The dyad was synthesized from meso-substituted trans-A2B-corrole bearing acid chloride functionality and meso-substituted A3B-porphyrin possessing one free NH2 group. The structure of the dyad was carefully designed and optimized to ensure stability of the molecule. The preparation of the amine component was achieved via phthalimide protection strategy which occurred to be more efficient than the traditional nitro group reduction. Results obtained from time resolved and steady state spectroscopy experiments indicate the existence of an equilibrium between the two lowest singlet excited states of the dyad, one localized on the corrole and the other on the porphyrin unit, which are nearly iso-energetic (ΔG = −0.01 eV). Independently of the excited component, energy transfer occurs in both directions (very likely with a Förster mechanism) and an equilibrium with Keq close to 1 is rapidly set with back and forward rates of the order of 109 s−1. Both states decay with a common lifetime (6.2 ns) that is longer compared to the corrole model (3.9 ns) and shorter with respect to the porphyrin reference (9.9 ns). The longer lived excited state localized on porphyrin acts as a reservoir for the excited state localized on corrole. At 77 K the equilibration does not take place during the lifetime of the excited states, and the decay of the two species occurs independently.Photoinduced processes in a new, stable corrole-porphyrin dyad made up of a free-base corrole and a free-base porphyrin connected by an amide linker have been determined. Independently of the excited component, an equilibrium mixture between the lowest excited singlet state localized on the two chromophores is established. The longer lived excited state localized on porphyrin acts as a reservoir for the excited state localized on corrole.
Co-reporter:Beata Koszarna, Holger Butenschön and Daniel T. Gryko
Organic & Biomolecular Chemistry 2005 vol. 3(Issue 14) pp:2640-2645
Publication Date(Web):16 Jun 2005
DOI:10.1039/B505366H
Ferrocene-bridged bisporphyrins have been synthesized by the condensation of corresponding dipyrromethane-derived diols with a bisdipyrromethane. Purification of the final compounds has been achieved without chromatography. The specific geometry of these bisporphyrins makes them valuable starting points for building complex molecular and supramolecular structures. In particular it provides a core to which multiple sites of attractive intermolecular interactions can be attached thereby creating compounds predisposed to form complex networks by association. We have studied the structure of bis-1,1′-(porphyrinyl)ferrocenes by 1H NMR, UV-Vis and electrochemistry. Results have shown that complex dynamic processes occur in these molecules (which may involve conformers, formation of H-aggregates and tautomers) and that they have non-typical electrochemical behaviour.
Co-reporter:Daniel T. Gryko;Beata Koszarna
European Journal of Organic Chemistry 2005 Volume 2005(Issue 15) pp:
Publication Date(Web):14 JUL 2005
DOI:10.1002/ejoc.200500089
The product distribution of the acid-mediated condensation of dipyrromethanes and aldehydes was studied and a novel type of macrocycle phlorin-dipyrrin conjugate was isolated and identified by X-ray analysis. The generality of its formation from sterically hindered dipyrromethanes and pentafluorophenyldipyrromethane was demonstrated. The influence of the meso-aryl groups on the stability of the phlorin was studied. The reported two-step synthesis of a phlorin derivative is one of the simplest routes leading to this type of molecules. (© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2005)
Co-reporter:Daniel T. Gryko and Beata Koszarna
Organic & Biomolecular Chemistry 2003 vol. 1(Issue 2) pp:350-357
Publication Date(Web):09 Dec 2002
DOI:10.1039/B208950E
We have refined a one-pot synthesis of A3-corroles via
“3+4” condensation of an aldehyde with a pyrrole followed by macrocyclization mediated by DDQ. After thorough examination of various reaction parameters (reactivity of an aldehyde, catalyst, solvent, concentration, time etc.) we have elaborated three different sets of conditions for different types of aromatic aldehydes—highly reactive, moderately reactive and sterically hindered. Thanks to the identification of the key factors influencing the yield of bilanes and the yield of their conversion to corroles we were able to improve yields to ca. 17% for highly reactive aldehydes and ca. 13% for moderately reactive aldehydes. Altogether fourteen A3-corroles have been prepared in 7–21% yield. 5,10,15-Trimesitylcorrole has been obtained for the first time. [2+1] Condensation between sterically hindered dipyrromethanes and aldehydes has also been refined and yields of trans-A2B-corroles have been improved by ca. 10%.
Co-reporter:Daniel T. Gryko
European Journal of Organic Chemistry 2002 Volume 2002(Issue 11) pp:
Publication Date(Web):8 MAY 2002
DOI:10.1002/1099-0690(200206)2002:11<1735::AID-EJOC1735>3.0.CO;2-K
The aim of this review is to highlight recent progress in the synthetic methodologies leading to corroles and core-modified corroles. Emphasis is put on corroles with meso substituents. (© Wiley-VCH Verlag GmbH, 69451 Weinheim, Germany, 2002)
Co-reporter:Beata Koszarna and Daniel T. Gryko
Chemical Communications 2007(Issue 28) pp:
Publication Date(Web):
DOI:10.1039/B703279J
Co-reporter:Lucia Flamigni and Daniel T. Gryko
Chemical Society Reviews 2009 - vol. 38(Issue 6) pp:NaN1646-1646
Publication Date(Web):2009/04/07
DOI:10.1039/B805230C
The collection and conversion of light energy into chemical energy is based on the use of molecular structures of various complexity, where the absorbed light energy is first converted into an excited state able to undergo energy or electron transfer processes and finally it is stored in a charge separated state as chemical (electrochemical) potential. A bio-mimetic approach has seen tetrapyrroles number among the most common components of these arrays. This tutorial review reports on the use of relatively new tetrapyrroles, corroles, in this field. A brief presentation of the electrochemical and photophysical properties of the corrole units relevant to the application is presented together with a discussion on the photo- and thermal stability issues, followed by an overview of the activity and improvements in the preparation of photo-active molecular arrays containing corroles.
Co-reporter:Lucia Flamigni, Dagmara Wyrostek, Roman Voloshchuk and Daniel T. Gryko
Physical Chemistry Chemical Physics 2010 - vol. 12(Issue 2) pp:NaN483-483
Publication Date(Web):2009/11/12
DOI:10.1039/B916525H
A dyad (C3–NI) based on corrole and naphthalene bisimide has been synthesized and its photoreactivity compared to that of the model component corrole (C3) and naphthalene bisimide (NI) in solvents of different polarity: toluene (TL) and dichloromethane (DCM). The major emitting species in NI solutions, in TL, is identified as a dimeric species (λ = 470 nm, τ = 2.3 ns) but traces of monomer can also be detected (λ ca. 390 nm, τ = 40 ps). In DCM the major emitting component is the monomer (λ = 383 nm, τ = 20 ps) but traces of different aggregates (λ = 540 and 570 nm, τ = 4.5 and 11 ns) are present. C3 has a fluorescence nearly unaffected by solvent polarity, with a maximum around 655 and a lifetime of 3.5 or 3.8 ns in DCM and TL, respectively. The dyad C3–NI does not appear to be affected by aggregation problems in any of the solvents. Excitation of the imide component in C3–NI (C3–1NI) results in an energy transfer to corrole (1C3–NI) with rate k = 2.0 × 1011 s−1 in both solvents. The latter state reacts further via a LUMO–LUMO electron transfer to the naphthalene bisimide yielding the charge separated state C3+–NI− (k = 1.8 × 109 s−1 in TL and k = 3.7 × 109 s−1 in DCM). The same type of reactivity is displayed by direct excitation of the corrole moiety in the dyad to 1C3–NI. C3+–NI− decays with a rate comparable to that of its formation in DCM (k = 4.0 × 109 s−1in DCM), precluding its accumulation, whereas it decays with a slower rate in TL (k = 7.1 × 108 s−1). The charge separated state recombines to the ground singlet state; recombination to the triplet state of corrole (excited state at the lowest energy) is in fact excluded on the basis of the experimentally determined triplet yields. The failure of the commonly used methods in the calculation of CS energy levels in apolar solvents is confirmed.
Co-reporter:Richard van Hameren, Johannes A. A. W. Elemans, Dagmara Wyrostek, Mariusz Tasior, Daniel T. Gryko, Alan E. Rowan and Roeland J. M. Nolte
Journal of Materials Chemistry A 2009 - vol. 19(Issue 1) pp:NaN69-69
Publication Date(Web):2008/10/17
DOI:10.1039/B812518J
The self-assembly of corrole trimers in solution and at solid–liquid interfaces is a process that depends on dewetting, hydrogen bonding and π–π interactions between the molecules forming columnar stacks, and lateral interactions between these stacks to generate higher order assemblies.
Co-reporter:Bartosz Bursa, Bolesław Barszcz, Waldemar Bednarski, Jan Paweł Lewtak, Dominik Koszelewski, Olena Vakuliuk, Daniel T. Gryko and Danuta Wróbel
Physical Chemistry Chemical Physics 2015 - vol. 17(Issue 11) pp:NaN7423-7423
Publication Date(Web):2015/02/04
DOI:10.1039/C4CP05648E
The investigation presented in this paper deals with new free-base corroles substituted with different peripheral groups. These aromatic macrocycles were efficiently synthesized by a [2+1] approach from dipyrromethanes. Moreover, the basic spectroscopic studies of the dyes in chloroform were conducted, and the UV-Vis absorption, fluorescence and ESR parameters were estimated. The experimental data were supported by quantum chemical calculations. The presence of monomeric dye structures is concentration independent (10−6–10−4 M), as expected for dyes in a solvent of low polarity, and rules out aggregate formation of corroles dissolved in chloroform. The excitation emission and fluorescence life-time values confirm the monomeric structure of the corroles. The spectra were compared with the time-dependent density functional theory (TD-DFT) results for the HOMO–LUMO states. The ESR examinations strongly show that for any type of studied fluorine corrole an unpaired electron is localized on the corrole macroring but not on the substituents both before and after light illumination. Laser illumination creates additional radicals, however with different effectiveness depending on the sample.