Co-reporter:Wahib Aggoune, Caterina Cocchi, Dmitrii Nabok, Karim Rezouali, Mohamed Akli Belkhir, and Claudia Draxl
The Journal of Physical Chemistry Letters April 6, 2017 Volume 8(Issue 7) pp:1464-1464
Publication Date(Web):March 15, 2017
DOI:10.1021/acs.jpclett.7b00357
By investigating the optoelectronic properties of prototypical graphene/hexagonal boron nitride (h-BN) heterostructures, we demonstrate how a nanostructured combination of these materials can lead to a dramatic enhancement of light–matter interaction and give rise to unique excitations. In the framework of ab initio many-body perturbation theory, we show that such heterostructures absorb light over a broad frequency range, from the near-infrared to the ultraviolet (UV), and that each spectral region is characterized by a specific type of excitations. Delocalized electron–hole pairs in graphene dominate the low-energy part of the spectrum, while strongly bound electron–hole pairs in h-BN are preserved in the near-UV. Besides these features, characteristic of the pristine constituents, charge-transfer excitations appear across the visible region. Remarkably, the spatial distribution of the electron and the hole can be selectively tuned by modulating the stacking arrangement of the individual building blocks. Our results open up unprecedented perspectives in view of designing van der Waals heterostructures with tailored optoelectronic features.
Co-reporter:Bernhard Klett, Caterina Cocchi, Linus Pithan, Stefan Kowarik and Claudia Draxl
Physical Chemistry Chemical Physics 2016 vol. 18(Issue 21) pp:14603-14609
Publication Date(Web):03 May 2016
DOI:10.1039/C6CP01405D
We present a joint theoretical and experimental study to investigate polymorphism in α-sexithiophene (6T) crystals. By means of density-functional theory calculations, we clarify that the low-temperature phase is favorable over the high-temperature one, with higher relative stability up to 50 meV per molecule. This result is in agreement with our thermal desorption measurements. We also propose a transition path between the high- and low-temperature 6T polymorphs, estimating an upper bound for the energy barrier of about 1 eV per molecule. The analysis of the electronic properties of the investigated 6T crystal structures complements our study.
Co-reporter:Christian Vorwerk, Caterina Cocchi, Claudia Draxl
Computer Physics Communications 2016 Volume 201() pp:119-125
Publication Date(Web):April 2016
DOI:10.1016/j.cpc.2016.01.004
Theoretical spectroscopy is a powerful tool to describe and predict optical properties of materials. While nowadays routinely performed, first-principles calculations only provide bulk dielectric tensors in Cartesian coordinates. These outputs are hardly comparable with experimental data, which are typically given by macroscopic quantities, crucially depending on the laboratory setup. Even more serious discrepancies can arise for anisotropic materials, e.g., organic crystals, where off-diagonal elements of the dielectric tensor can significantly contribute to the spectral features. Here, we present LayerOptics, a versatile and user-friendly implementation, based on the solution of the Maxwell’s equations for anisotropic materials, to compute optical coefficients in anisotropic layered materials. We apply this tool for post-processing full dielectric tensors of molecular materials, including excitonic effects, as computed from many-body perturbation theory using the exciting code. For prototypical examples, ranging from optical to X-ray frequencies, we show the importance of combining accurate ab initio methods to obtain dielectric tensors, with the solution of the Maxwell’s equations to compute optical coefficients accounting for optical anisotropy of layered systems. Good agreement with experimental data supports the potential of our approach, in view of achieving microscopic understanding of spectroscopic properties in complex materials.
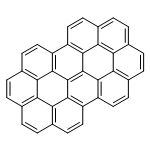
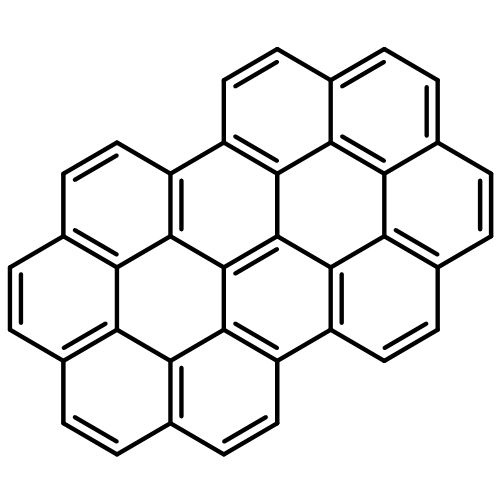
Co-reporter:Caterina Cocchi, Deborah Prezzi, Alice Ruini, Marilia J. Caldas, and Elisa Molinari
The Journal of Physical Chemistry A 2014 Volume 118(Issue 33) pp:6507-6513
Publication Date(Web):July 1, 2014
DOI:10.1021/jp503054j
The electronic and optical properties of polycyclic aromatic hydrocarbons (PAHs) present a strong dependence on their size and geometry. We tackle this issue by analyzing the spectral features of two prototypical classes of PAHs, belonging to D6h and D2h symmetry point groups and related to coronene as multifunctional seed. While the size variation induces an overall red shift of the spectra and a redistribution of the oscillator strength between the main peaks, a lower molecular symmetry is responsible for the appearance of new optical features. Along with broken molecular orbital degeneracies, optical peaks split and dark states are activated in the low-energy part of the spectrum. Supported by a systematic analysis of the composition and the character of the optical transitions, our results contribute in shedding light to the mechanisms responsible for spectral modifications in the visible and near UV absorption bands of medium-size PAHs.
Co-reporter:Bernhard Klett, Caterina Cocchi, Linus Pithan, Stefan Kowarik and Claudia Draxl
Physical Chemistry Chemical Physics 2016 - vol. 18(Issue 21) pp:NaN14609-14609
Publication Date(Web):2016/05/03
DOI:10.1039/C6CP01405D
We present a joint theoretical and experimental study to investigate polymorphism in α-sexithiophene (6T) crystals. By means of density-functional theory calculations, we clarify that the low-temperature phase is favorable over the high-temperature one, with higher relative stability up to 50 meV per molecule. This result is in agreement with our thermal desorption measurements. We also propose a transition path between the high- and low-temperature 6T polymorphs, estimating an upper bound for the energy barrier of about 1 eV per molecule. The analysis of the electronic properties of the investigated 6T crystal structures complements our study.