Co-reporter:Ismail M. Taban, Jinge Zhu, Hector F. DeLuca, Claire Simons
Bioorganic & Medicinal Chemistry 2017 Volume 25, Issue 20(Issue 20) pp:
Publication Date(Web):15 October 2017
DOI:10.1016/j.bmc.2017.08.036
A homology model of human CYP27B1 was built using MOE and was further optimised by molecular dynamics simulations of the hCYP27B1 homology model and a hCYP27B1-SDZ-88357 complex. Docking results from the hCYP27B1-SDZ-88357 complex showed amino acids Arg107, Asn387 and Asp320 have an important role in binding interaction, with Asp320 part of the important acid-alcohol pair situated in the I-helix with the conserved sequence (A/G) GX (E/D) (T/S), which assumes an essential role in the binding of an oxygen molecule for catalysis. Additional docking experiments with selective hCYP27B1 or hCYP24A1 inhibitors using both the hCYP27B1 model and a triple mutant hCYP24A1 model provided further support for the importance of H-bonding interactions with the three identified active site amino acids. To confirm the role of Arg107, Asn387 and Asp320 in the active site of hCYP27B1 compounds were designed that would form H-bonding interactions, as determined from docking experiments with the hCYP27B1 model. Subsequent synthesis and CYP24A1 and CYP27B1 enzyme assays of the designed compounds 1a and 1b showed a ∼5-fold selectivity for CYP27B1 confirming the importance of Asp320 in particular and also Asn387 and Arg107 as important amino acids for CYP27B1 inhibitory activity.Download high-res image (75KB)Download full-size image
Co-reporter:Ismail M. Taban, Jinge Zhu, Hector F. DeLuca, Claire Simons
Bioorganic & Medicinal Chemistry 2017 Volume 25, Issue 15(Issue 15) pp:
Publication Date(Web):1 August 2017
DOI:10.1016/j.bmc.2017.05.055
CYP24A1 (25-hydroxyvitamin D-24-hydroxylase) is a useful enzyme target for a range of medical conditions including cancer, cardiovascular and autoimmune disease, which show elevated CYP24A1 levels and corresponding reduction of calcitriol (the biologically active form of vitamin D). A series of (E)-N-(2-(1H-imidazol-1-yl)-2-(phenylethyl)-3/4-styrylbenzamides have been synthesised using an efficient synthetic route and shown to be potent inhibitors of CYP24A1 (IC50 0.11–0.35 μM) compared with the standard ketoconazole. Molecular modelling using our CYP24A1 homology model showed the inhibitors to fill the hydrophobic binding site, forming key transition metal interaction between the imidazole nitrogen and the haem Fe3+ and multiple interactions with the active site amino acid residues.Download high-res image (113KB)Download full-size image
Co-reporter:Salvatore Ferla ; Ahmed S. Aboraia ; Andrea Brancale ; Christopher J. Pepper ; Jinge Zhu ; Justin T. Ochalek ; Hector F. DeLuca
Journal of Medicinal Chemistry 2014 Volume 57(Issue 18) pp:7702-7715
Publication Date(Web):August 22, 2014
DOI:10.1021/jm5009314
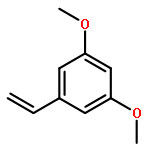
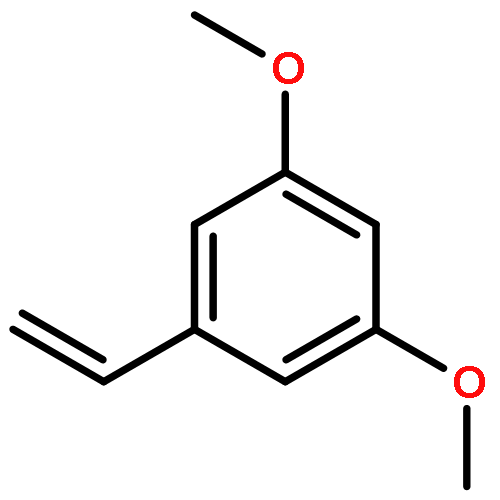
The synthesis of imidazole styrylbenzamide, tert-butyl styrylimidazole, and tert-butyl styrylsulfonate derivatives is described. Evaluation of binding affinity and inhibitory activity against CYP24A1 identified the imidazole styrylbenzamides as potent inhibitors of CYP24A1, having selectivity with respect to CYP27B1 comparable with or greater than that of the standard ketoconazole. Further evaluation of the 3,5-dimethoxy and 3,4,5-trimethoxy derivatives in chronic lymphocytic leukemia cells revealed that co-treatment of 1α,25-dihydroxyvitamin D3 plus inhibitor coordinately upregulated GADD45α and CDKN1A. Docking experiments on the inhibitors in the CYP24A1 enzyme active site suggest the compounds reach the active site through the vitamin D access tunnel and are exposed to multiple hydrophobic residues. The imidazole styrylbenzamides are optimally positioned to allow interaction of the imidazole with the heme, and, in the case of the methoxy derivatives, a hydrogen bond between the 3-methoxy group and Gln82 stabilizes the molecule in a favorable active conformation.
Co-reporter:Salvatore Ferla, Mohamed S. Gomaa, Andrea Brancale, Jinge Zhu, Justin T. Ochalek, Hector F. DeLuca, Claire Simons
European Journal of Medicinal Chemistry 2014 Volume 87() pp:39-51
Publication Date(Web):24 November 2014
DOI:10.1016/j.ejmech.2014.09.035
•New series of imidazole styrylindoles and sulfonyl styrylindoles synthesized.•Compounds evaluated as inhibitors of 25-hydroxyvitamin D-24-hydroxylase (CYP24A1).•Imidazole series potent CYP24A1 inhibitors.•Computational studies identified key enzyme binding interactions.The synthesis of a series of imidazole styrylindoles and sulfonyl styrylindoles derivatives is described. Evaluation of binding affinity and inhibitory activity against CYP24A1 identified the imidazole styrylindoles as potent inhibitors with activity greater or comparable with the standard ketoconazole.Flexible alignment and docking studies of the inhibitors in the CYP24A1 enzyme active site confirmed that complete occupation of the vitamin D access tunnel is essential to inhibitory activity, allowing exposure to multiple hydrophobic binding interactions and optimal conformation for the interaction of the imidazole nitrogen lone pair and the active site haem.A series of imidazole styrylindoles and sulfonyl styrylindoles derivatives were prepared and evaluated for their binding affinity and inhibitory activity against CYP24A1 with molecular modelling studies to determine key binding interactions within the CYP24A1 active site.
Co-reporter:Mohamed S. Gomaa, Caroline E. Bridgens, Nicola A. Illingworth, Gareth J. Veal, Christopher P.F. Redfern, Andrea Brancale, Jane L. Armstrong, Claire Simons
Bioorganic & Medicinal Chemistry 2012 Volume 20(Issue 14) pp:4201-4207
Publication Date(Web):15 July 2012
DOI:10.1016/j.bmc.2012.05.076
Retinoic acid (RA), the biologically active metabolite of vitamin A, is used medicinally for the treatment of hyperproliferative diseases including dermatological conditions and cancer. The antiproliferative effects of RA have been well documented as well as the limitations owing to toxicity and the development of resistance to RA therapy. RA metabolism inhibitors (RAMBAs or CYP26 inhibitors) are attracting increasing interest as an alternative method for enhancing endogenous levels of retinoic acid in the treatment of hyperproliferative disease. Here the synthesis and inhibitory activity of novel 3-(1H-imidazol- and triazol-1-yl)-2,2-dimethyl-3-(4-(phenylamino)phenyl)propyl derivatives in a MCF-7 CYP26A1 microsomal assay are described. The most promising inhibitor methyl 2,2-dimethyl-3-(4-(phenylamino)phenyl)-3-(1H-1,2,4-triazol-1-yl)propanoate (6) exhibited an IC50 of 13 nM (compared with standards Liarozole IC50 540 nM and R116010 IC50 10 nM) and was further evaluated for CYP selectivity using a panel of CYP with >100-fold selectivity for CYP26 compared with CYP1A2, 2C9 and 2D6 observed and 15-fold selectivity compared with CYP3A4. The results demonstrate the potential for further development of these potent inhibitors.The synthesis and inhibitory activity of novel 3-(1H-imidazol- and triazol-1-yl)-2,2-dimethyl-3-(4-(phenylamino)phenyl)propyl derivatives in a MCF-7 CYP26A1 microsomal assay are described. The most promising inhibitor methyl 2,2-dimethyl-3-(4-(phenylamino)phenyl)-3-(1H-1,2,4-triazol-1-yl)propanoate (6, R = H, X = N) exhibited an IC50 of 13 nM (compared with standards Liarozole IC50 540 nM and R116010 IC50 10 nM) and >100-fold selectivity for CYP26 compared with CYP1A2, 2C9 and 2D6 observed and 15-fold selectivity compared with CYP3A4. The results demonstrate the potential for further development of these potent inhibitors.
Co-reporter:Mohamed S. Gomaa, Andrew S.T. Lim, S.C. Wilson Lau, Ann-Marie Watts, Nicola A. Illingworth, Caroline E. Bridgens, Gareth J. Veal, Christopher P.F. Redfern, Andrea Brancale, Jane L. Armstrong, Claire Simons
Bioorganic & Medicinal Chemistry 2012 Volume 20(Issue 20) pp:6080-6088
Publication Date(Web):15 October 2012
DOI:10.1016/j.bmc.2012.08.044
The role of all-trans-retinoic acid (ATRA) in the development and maintenance of many epithelial and neural tissues has raised great interest in the potential of ATRA and related compounds (retinoids) as pharmacological agents, particularly for the treatment of cancer, skin, neurodegenerative and autoimmune diseases. The use of ATRA or prodrugs as pharmacological agents is limited by a short half-life in vivo resulting from the activity of specific ATRA hydroxylases, CYP26 enzymes, induced by ATRA in liver and target tissues. For this reason retinoic acid metabolism blocking agents (RAMBAs) have been developed for treating cancer and a wide range of other diseases.The synthesis, CYP26A1 inhibitory activity and molecular modeling studies of novel methyl 3-[4-(arylamino)phenyl]-3-(azole)-2,2-dimethylpropanoates are presented. From this series of compounds clear SAR can be derived for 4-substitution of the phenyl ring with electron-donating groups more favourable for inhibitory activity. Both the methylenedioxyphenyl imidazole (17, IC50 = 8 nM) and triazole (18, IC50 = 6.7 nM) derivatives were potent inhibitors with additional binding interactions between the methylenedioxy moiety and the CYP26 active site likely to be the main factor. The 6-bromo-3-pyridine imidazole 15 (IC50 = 5.7 nM) was the most active from this series compared with the standards liarozole (IC50 = 540 nM) and R116010 (IC50 = 10 nM).A series of 3-[4-(arylamino)phenyl]-3-(azole)-2,2-dimethylpropanoates were prepared and evaluated for their inhibitory activity against CYP26A1 with molecular modeling studies to determine key binding interactions within the CYP26A1 active site.
Co-reporter:Afraa Siam;Andrea Brancale
Journal of Molecular Modeling 2012 Volume 18( Issue 2) pp:441-453
Publication Date(Web):2012 February
DOI:10.1007/s00894-011-1084-6
CYP7B1 mutations have been linked directly with the neurodegenerative disease hereditary spastic paraplegia (HSP), with mutations in the CYP7B1 gene identified as being directly responsible for autosomal recessive HSP type 5A (SPG5). To evaluate the potential impact of CYP7B1 mutations identified in SPG5 on binding and protein function, a comparative model of cytochrome P450 7B1 (CYP7B1) was constructed using human CYP7A1 as a template during model construction. The secondary structure was predicted using the PSIPRED and GOR4 prediction methods, the lowest energy CYP7B1 model was generated using MOE, and then this model was assessed in terms of stereochemical quality and the side chain environment using RAMPAGE, Verify3D and ProSA. Evaluation of the active site residues of the CYP7B1 model and validation of the active site architecture were performed via molecular docking experiments: the docking of the substrates 25-hydroxycholesterol and 27-hydroxycholesterol and the inhibitor 3α-Adiol identified structurally and functionally important residues. Mutational analysis of CYP7B1 amino acid mutations related to hereditary spastic paraplegia type 5 considered phosphorylation, ligand/substrate binding and the structural roles of mutated amino acid residues, with R112, T297 and S363 mutations expected to have a direct impact on ligand binding, while mutations involving R417 would indirectly affect ligand binding as a result of impairment in catalytic function.
Co-reporter:Mohamed S. Gomaa ; Caroline E. Bridgens ; Ahmed S. Aboraia ; Gareth J. Veal ; Christopher P. F. Redfern ; Andrea Brancale ; Jane L. Armstrong
Journal of Medicinal Chemistry 2011 Volume 54(Issue 8) pp:2778-2791
Publication Date(Web):March 23, 2011
DOI:10.1021/jm101583w
The synthesis and potent inhibitory activity of novel imidazole methyl 3-(4-(aryl-2-ylamino)phenyl)propanoates in a MCF-7 CYP26A1 microsomal assay is described. The induction of CYP26A1 mRNA was used to evaluate the ability of the compounds to enhance the biological effects of all-trans retinoic acid (ATRA) in a retinoid-responsive neuroblastoma cell line. The most promising inhibitor, 3-imidazol-1-yl-2-methyl-3-[4-(naphthalen-2-ylamino)-phenyl]-propionic acid methyl ester (20), with an IC50 of 3 nM (compared with liarozole IC50 of 540 nM and R116010 IC50 of 10 nM) was further evaluated for CYP selectivity using a panel of CYP enzymes, mutagenicity (Ames screen), and hepatic stability.
Co-reporter:Mohamed S. Gomaa ; Caroline E. Bridgens ; Gareth J. Veal ; Christopher P. F. Redfern ; Andrea Brancale ; Jane L. Armstrong
Journal of Medicinal Chemistry 2011 Volume 54(Issue 19) pp:6803-6811
Publication Date(Web):August 15, 2011
DOI:10.1021/jm200695m
The synthesis and potent inhibitory activity of novel 3-(1H-imidazol- and triazol-1-yl)-2,2-dimethyl-3-(4-(naphthalen-2-ylamino)phenyl)propyl derivatives vs a MCF-7 CYP26A1 microsomal assay is described. This study focused on the effect of modifying the heme binding azole group and the flexible C3 chain on inhibitory activity and selectivity. The most promising inhibitor 2,2-dimethyl-3-[4-(naphthalen-2-ylamino)-phenyl]-3-[1,2,4]triazol-1-yl-propionic acid methyl ester (17) (IC50 = 0.35 nM as compared with liarozole IC50 = 540 nM and R116010 IC50 = 10 nM) was evaluated for CYP selectivity and hepatic stability. Compounds with CYP26 inhibitory IC50 values ≤50 nM enhanced the biological activity of exogenous ATRA, as evidenced by a 3.7–5.8-fold increase in CYP26A1 mRNA in SH-SY5Y neuroblastoma cells as compared with ATRA alone. All compounds demonstrated an activity comparable with or better than R116010, and the induction correlated well with CYP26 inhibition data. These studies highlight the promising activity profile of this novel CYP26 inhibitor and suggest it as an appropriate candidate for future development.
Co-reporter:Ehab Al-Moubarak
Journal of Molecular Modeling 2011 Volume 17( Issue 7) pp:1679-1693
Publication Date(Web):2011 July
DOI:10.1007/s00894-010-0871-9
Treatment of C. difficile infection is one of the most difficult biomedical challenges. To develop novel antibacterials, researchers have been targeting bacterial molecular functions that are essential for its growth. The methionyl tRNA synthetase (MetRS) is strictly required for protein biosynthesis and success was reported in developing antibacterials to inhibit this enzyme. The present study was aimed at building and analyzing a homology model for C. difficile MetRS in the context of drug design. A homology model of C. difficile MetRS was constructed using Molecular Operating Environment (MOE) software. A. aeolicus MetRS was the main template while the query zinc binding domain was modeled using T. thermophilus MetRS. The model has been assessed and compared to its main template (Ramachandran, ERRAT and ProSA). The active site of the query protein has been predicted from its sequence using a detailed conservation analysis (ClustalW2). Using MOE software, suitable ligands were docked in the constructed model, including a C. difficile MetRS inhibitor REP3123 and the enzyme natural substrate, and the key active site residues and interactions were identified. These docking studies have validated the active site conformation in the constructed model and identified binding interactions.
Co-reporter:Ahmed S. Aboraia, Bart Makowski, Alba Bahja, David Prosser, Andrea Brancale, Glenville Jones, Claire Simons
European Journal of Medicinal Chemistry 2010 Volume 45(Issue 10) pp:4427-4434
Publication Date(Web):October 2010
DOI:10.1016/j.ejmech.2010.07.001
A series of (E)-2-(2-substituted benzylidene)- and 2-(2-substituted benzyl)-6-methoxy-tetralones were prepared, using an efficient synthetic scheme, and evaluated for their inhibitory activity against cytochrome P450C24A1 (CYP24A1) hydroxylase. In general the reduced benzyl tetralones were more active than the parent benzylidene tetralones. The 2-ethyl and 2-trifluoromethyl benzyl tetralone derivatives (4c and 4b) showed optimal activity in this series with IC50 values of 1.92 μM and 2.08 μM, respectively compared with the standard ketoconazole IC50 0.52 μM. The 2-bromobenzyl tetralone (4d) showed a preference for CYP27A1 (IC50 59 nM) over CYP24A1 (IC50 16.3 μM) and may be a useful lead in CYP27A1 inhibition studies. The 2-ethylphenyl benzyl derivative (9c), which showed weak activity against the wild type CYP24A1 (IC50 25.57 μM), exhibited enhanced inhibitory activity towards L148F and M416T mutants, this difference in activity for the L148F mutant has been explained using molecular modelling.A series of (E)-2-(2-substituted benzylidene)- and 2-(2-substituted benzyl)-6-methoxy-tetralones were prepared and evaluated for their inhibitory activity against cytochrome CYP24A1, CYP27A1 and CYP24A1 mutant strains.
Co-reporter:Ahmed S. Aboraia, Sook Wah Yee, Mohamed Sayed Gomaa, Nikhil Shah, Anna C. Robotham, Bart Makowski, David Prosser, Andrea Brancale, Glenville Jones, Claire Simons
Bioorganic & Medicinal Chemistry 2010 Volume 18(Issue 14) pp:4939-4946
Publication Date(Web):15 July 2010
DOI:10.1016/j.bmc.2010.06.011
A series of N-(2-(1H-imidazol-1-yl)-2-phenylethyl)arylamides were prepared, using an efficient three- to five-step synthesis, and evaluated for their inhibitory activity against human cytochrome P450C24A1 (CYP24A1) hydroxylase. Inhibition ranged from IC50 0.3–72 μM compared with the standard ketoconazole IC50 0.52 μM, with the styryl derivative (11c) displaying enhanced activity (IC50 = 0.3 μM) compared with the standard, providing a useful preliminary lead for drug development.A series of N-(2-(1H-imidazol-1-yl)-2-phenylethyl)arylamides were prepared and evaluated for their inhibitory activity against human cytochrome P450C24A1 (CYP24A1) hydroxylase. The styryl derivative (11c) displayed enhanced activity (IC50 = 0.3 μM) compared with the standard ketoconazole, providing a useful lead.
Co-reporter:Mohamed Sayed Gomaa, Jane L. Armstrong, Beatrice Bobillon, Gareth J. Veal, Andrea Brancale, Christopher P.F. Redfern, Claire Simons
Bioorganic & Medicinal Chemistry 2008 Volume 16(Issue 17) pp:8301-8313
Publication Date(Web):1 September 2008
DOI:10.1016/j.bmc.2007.06.048
The synthesis and potent inhibitory activity of novel 4-[(imidazol-1-yl and triazol-1-yl)(phenyl)methyl]aryl-and heteroaryl amines versus a MCF-7 CYP26A1 cell assay is described. Biaryl imidazole ([4-(imidazol-1-yl-phenyl-methyl)-phenyl]-naphthalen-2-yl-amine (8), IC50 = 0.5 μM; [4-(imidazol-1-yl-phenyl-methyl)-phenyl]-indan-5-yl-amine (9), IC50 = 1.0 μM) and heteroaryl imidazole derivatives ((1H-benzoimidazol-2-yl)-{4-[(5H-imidazol-1-yl)-phenyl-methyl]-phenyl}-amine (15), IC50 = 2.5 μM; benzooxazol-2-yl-{4-[(5H-imidazol-1-yl)-phenyl-methyl]-phenyl}-amine (16), IC50 = 0.9 μM; benzothiazol-2-yl-{4-[(5H-imidazol-1-yl)-phenyl-methyl]-phenyl}-amine (17), IC50 = 1.5 μM) were the most potent CYP26 inhibitors. Using a CYP26A1 homology model differences in activity were investigated. Incubation of SH-SY5Y human neuroblastoma cells with the imidazole aryl derivative 8, and the imidazole heteroaryl derivatives 16 and 17 potentiated the atRA-induced expression of CYP26B1. These data suggest that further structure–function studies leading to clinical development are warranted.
Co-reporter:Stephane Pautus, Sook Wah Yee, Martyn Jayne, Michael P. Coogan, Claire Simons
Bioorganic & Medicinal Chemistry 2006 Volume 14(Issue 11) pp:3643-3653
Publication Date(Web):1 June 2006
DOI:10.1016/j.bmc.2006.01.018
Methodology previously described by our group was applied to the preparation of a series of 4-alkyl/aryl-substituted 1-[benzofuran-2-yl-phenylmethyl]-1H-triazoles. The [1,2,4]-triazole derivatives were prepared for a range of alkyl and aryl substituents, and for the 4-methyl, 4-ethyl, 4-ipropyl, 4-tbutyl, 4-phenyl and 4-chlorophenyl derivatives, the minor [1,3,4]-triazole isomer also isolated. All the triazole derivatives were evaluated for CYP26A1 inhibitory activity using a MCF-7 cell-based assay. The 4-ethyl and 4-phenyl-1,2,4-triazole derivatives displayed inhibitory activity (IC50 4.5 and 7 μM, respectively) comparable with that of the CYP26 inhibitor liarozole (IC50 7 μM). Using a CYP26A1 homology model (based on CYP3A4) template, docking experiments were performed with MOE with multiple hydrophobic interactions observed in addition to coordination between the triazole nitrogen and the haem transition metal.A series of 4-alkyl/aryl-substituted 1-[benzofuran-2-yl-phenylmethyl]-1H-[1,2,4] and [1,3,4]triazoles derivatives were prepared and evaluated for CYP26A1 inhibitory activity using a MCF-7 cell based assay. The 4-ethyl and 4-phenyl-1,2,4-triazole derivatives displayed inhibitory activity (IC50 4.5 and 7 μM, respectively) comparable with the CYP26 inhibitor liarozole (IC50 7 μM). Using a CYP26A1 homology model, docking experiments were performed with the inhibitor compounds.
Co-reporter:Nurolaini Kifli, Thet Thet Htar, Erik De Clercq, Jan Balzarini, Claire Simons
Bioorganic & Medicinal Chemistry 2004 Volume 12(Issue 12) pp:3247-3257
Publication Date(Web):15 June 2004
DOI:10.1016/j.bmc.2004.03.072
Methodology previously described by us was applied to the formation of novel conformationally restrained bicyclic sugar modified nucleosides, with introduction of an oxazole and a thiocarbamate ring at the 2′,3′-positions of the ribonucleosides. Two novel alkyl derivatives of 2′,3′-dideoxy-2′,3′-oxazole-β-d-uridine and a novel uridine 2′,3′-thiocarbamate were successfully synthesised. Conformational evaluation of all the synthesised compounds was conducted using the theoretical potential energy calculation via the macromodel v.6.0 molecular modelling programme. The conformationally restrained nucleosides described were evaluated against a wide range of DNA and RNA viruses. None of the compounds showed specific antiviral effects at subtoxic concentrations.Two novel conformationally restrained alkyl derivatives of 2′,3′-dideoxy-2′,3′-oxazole-β-d-uridine and a novel uridine 2′,3′-thiocarbamate were prepared and conformational parameters determined using macromodel v.6.0 molecular modelling programme. The novel nucleosides were evaluated against a wide range of viral types and strains in cell culture.
Co-reporter:Nurolaini Kifli, Erik De Clercq, Jan Balzarini, Claire Simons
Bioorganic & Medicinal Chemistry 2004 Volume 12(Issue 15) pp:4245-4252
Publication Date(Web):1 August 2004
DOI:10.1016/j.bmc.2004.05.017
The preparation of a series of novel 6-(β-D-ribofuranosyl)-2-alkyl/aryl-6H-imidazo[1,2-c]pyrimidin-5-one nucleosides and the 2-nitrile nucleosides, 6-(β-D-ribofuranosyl)-5-oxo-5,6-dihydro-imidazo[1,2-c]pyrimidine-2-carbonitrile and 2R and 2S isomers of 6-(β-D-ribofuranosyl)-5-oxo-2,3,5,6-tetrahydro-imidazo[1,2-c]pyrimidine-2-carbonitrile, is described using two synthetic approaches. The nucleoside mimetics described were evaluated against a wide range of viral types and strains in cell culture. With the exception of one nucleoside, which displayed anti-CMV activity at toxic concentrations, none of the compounds showed antiviral activity most likely due to a lack of substrate recognition by viral and/or cellular nucleoside kinases.Novel 6-(β-D-ribofuranosyl)-2-alkyl/aryl-6H-imidazo[1,2-c]pyrimidin-5-one and 6-(β-D-ribofuranosyl)-5-oxo-5,6-dihydro- and 2,3,5,6-tetrahydro-imidazo[1,2-c]pyrimidine-2-carbonitrile nucleosides were prepared and evaluated against a wide range of viral types and strains in cell culture.
Co-reporter:Samar S. Elbaramawi, Samy M. Ibrahim, El-Sayed M. Lashine, Mohamed E. El-Sadek, Efi Mantzourani, Claire Simons
Journal of Molecular Graphics and Modelling (May 2017) Volume 73() pp:
Publication Date(Web):1 May 2017
DOI:10.1016/j.jmgm.2017.02.002
•A three-dimensional structural model of S.aureus PheRS has been constructed.•Catalytic, magnesium and tRNA binding sites have been analysed.•Analysis of substrate and inhibitor binding interactions through molecular docking.•Key amino acid interactions at binding sites identified for inhibitor design.Increased resistance of MRSA (multidrug resistance Staphylococcus aureus) to anti-infective drugs is a threat to global health necessitating the development of anti-infectives with novel mechanisms of action. Phenylalanine tRNA synthetase (PheRS) is a unique enzyme of the aminoacyl-tRNA synthetases (aaRSs), which are essential enzymes for protein biosynthesis. PheRS is an (αb)2 tetrameric enzyme composed of two alpha subunits (PheS) and two larger beta subunits (PheT). Our potential target in the drug development for the treatment of MRSA infections is the phenylalanine tRNA synthetase alpha subunit that contains the binding site for the natural substrate. There is no crystal structure available for S. aureus PheRS, therefore comparative structure modeling is required to establish a putative 3D structure for the required enzyme enabling development of new inhibitors with greater selectivity. The S. aureus PheRS alpha subunit homology model was constructed using Molecular Operating Environment (MOE) software. Staphylococcus haemolyticus PheRS was the main template while Thermus thermophilus PheRS was utilised to predict the enzyme binding with tRNAphe. The model has been evaluated and compared with the main template through Ramachandran plots, Verify 3D and Protein Statistical Analysis (ProSA). The query protein active site was predicted from its sequence using a conservation analysis tool. Docking suitable ligands using MOE into the constructed model were used to assess the predicted active sites. The docked ligands involved the PheRS natural substrate (phenylalanine), phenylalanyl-adenylate and several described S. aureus PheRS inhibitors.Download high-res image (171KB)Download full-size image

![Ferrate(2-), [7,12-diethenyl-3,8,13,17-tetramethyl-21H,23H-porphine-2,18-dipropanoato(4-)-κN21,κN22,κN23,κN24]-, hydrogen (1:2), (SP-4-2)-](/data/chemimg/522800/14875-96-8.png)
![Ferrate(2-), [7,12-diethenyl-3,8,13,17-tetramethyl-21H,23H-porphine-2,18-dipropanoato(4-)-κN21,κN22,κN23,κN24]-, hydrogen (1:2), (SP-4-2)-](/data/chemimg/522800/14875-96-8_b.png)

![Benzoic acid, 4-[(1E)-2-phenylethenyl]-](http://img.cochemist.com/ccimg/13100/13041-75-3.png)
![Benzoic acid, 4-[(1E)-2-phenylethenyl]-](http://img.cochemist.com/ccimg/13100/13041-75-3_b.png)