Co-reporter:Maik Grätz;Andreas Bäcker;Lisa Vondung;Leon Maser;Arian Reincke
Chemical Communications 2017 vol. 53(Issue 53) pp:7230-7233
Publication Date(Web):2017/06/29
DOI:10.1039/C7CC02335A
We report a novel method for the preparation of PBP-pincer complexes from bis(phosphine)boronium salts. The central (R3P)2HB-moiety in a palladium complex is demonstrated to be a L-type ligand, therewith completing a series of pincer-type complexes with Z-, X- and L-type boron-based ligands, respectively.
Co-reporter:Felix Schneck, Maik Assmann, Markus Balmer, Klaus Harms, and Robert Langer
Organometallics 2016 Volume 35(Issue 11) pp:1931-1943
Publication Date(Web):May 23, 2016
DOI:10.1021/acs.organomet.6b00251
A comparative study on the synthesis, stability, and catalytic activity of various iron pincer complexes with the general formula [(R-PNHP)Fe(H) (CO) (BH4)] is reported, where R denotes the substituent of the terminal PR2-groups (R = tBu, Cy, iPr, Ph, Et). By the example of the synthesized precatalysts, it is shown that the nature of the ligands has a surprising influence on the catalytic properties of the complexes. Bulky ligands and less electron donating ligands affect the stability of the complexes, which preferably react under the loss of CO or H2 to deactivated products. In return, the reduced steric demand and the strong σ-donating properties of the Et-substituted precatalyst (2a) lead to an improved activity in the hydrogenation of esters to alcohols, compared to that of the previously reported iPr-substituted complexes. The improved activity of complex 2a is clearly demonstrated in the direct hydrogenation of amides to alcohols and amines under mild conditions.
Co-reporter:W. Xu and R. Langer
Dalton Transactions 2015 vol. 44(Issue 38) pp:16785-16790
Publication Date(Web):18 Aug 2015
DOI:10.1039/C5DT02226F
A series of ruthenium(II) complexes with three novel PNX-type pincer ligands is reported, in which X denotes a heterocyclic donor group (PNX = Ph2PCH2CH2N(H)CH2-X, X = 2-pyridyl, 2-furanyl, 2-thiophenyl or 2-pyrrolyl). The reaction of [(Ph3P)3RuCl2] with one equivalent of ligand leads to the trans-dichloro complexes [(PNX)RuCl2(PPh3)] (1–4) for all ligands, whereas the complexation of [(Ph3P)3Ru(H)(Cl)(CO)] results in different types of complexes. The variation of the heterocyclic donor group is in line with different binding properties and a labilization of this group. Investigations on the catalytic activity of different types of ruthenium(II) complexes in hydrogenation and dehydrogenation reactions reveal that more labile bound heterocycle donors result in a decrease of catalytic productivity and activity.
Co-reporter:Robert Langer;Friedrich Bönisch;Leon Maser;Clemens Pietzonka;Lisa Vondung;Thomas Philipp Zimmermann
European Journal of Inorganic Chemistry 2015 Volume 2015( Issue 1) pp:141-148
Publication Date(Web):
DOI:10.1002/ejic.201402859
Abstract
A series of iron(II) dihalogenide complexes with two different bisphosphinoethane ligands is reported. In the case of 1,2-bis(diphenylphosphanyl)ethane (dppe), depending on the stoichiometry, the tetrahedral [(μ-dppe)FeCl2]n and octahedral trans-[(dppe)2FeCl2] complexes are formed. The polymeric complex [(μ-dppe)FeCl2]n, with iron in a tetrahedral environment, preferentially reacts with chelating amines to give the octahedral diphosphine complex, trans-[(dppe)2FeCl2], and different octahedral amine complexes. With the sterically more demanding 1,2-bis(diisopropylphosphanyl)ethane (dippe), the monomeric and tetrahedral halogen complexes [(dippe)FeX2] are exclusively obtained (X = Cl, Br). These complexes react with chelating amines in a similar manner, to give free ligand and the corresponding octahedral amine complex. The present results suggest that the diphosphines in the investigated iron(II) complexes are bound too weakly to form productive catalyst precursors.
Co-reporter:Robert Langer;Alexer Gese;Donatas Gesevi&x10d;ius;Maximilian Jost;Bastian R. Langer;Felix Schneck;Alexer Venker;Weiqin Xu
European Journal of Inorganic Chemistry 2015 Volume 2015( Issue 4) pp:696-705
Publication Date(Web):
DOI:10.1002/ejic.201402970
Abstract
The synthesis, reactivity, and catalytic activity of RuII complexes with different pyridine- and imidazole-based P,N ligands are reported. The investigations reveal a strong influence of the N-heterocycle and the steric demand of the phosphine groups on the stability of different isomers of [L2RuX2] (L = P,N ligand; X = Cl, H). The imidazole-based complex 5 with dicyclohexylphosphine groups was found to be the most active precatalyst for the acceptorless dehydrogenation of primary alcohols, whereas different phosphine groups at the imidazole ligand as well as pyridine-based ligands caused a drop in catalytic activity. In the presence of a primary amine, imines are preferentially formed under these conditions. In summary, the investigations show that comparably small changes in the ligand moiety have a strong effect on the relative stability of stereoisomers for both hydride and chloride complexes, whereas isomerization of the kinetic reaction products was observed in some cases. The described changes in the ligand moiety most probably have a strong impact on the relative stabilities of isomeric intermediates as well and thus affect the catalytic activity of these complexes.
Co-reporter:Nicolas Frank, Katharina Hanau, and Robert Langer
Inorganic Chemistry 2014 Volume 53(Issue 20) pp:11335-11343
Publication Date(Web):October 7, 2014
DOI:10.1021/ic5022164
The octahedral transition-metal complex [(dppa)Fe(Ph2P–N–PPh2)2] (1) [dppa = bis(diphenylphosphino)amine] with homofunctional bidentate ligands is described. The ligand exhibits hemilability due to its small bite angle and the steric repulsion of the coordinated donor groups. As the {Ph2P–N–PPh2}− ligand can act as an internal base, heterolytic cleavage of dihydrogen by complex 1 leads to the formation of the hydride complex [(dppa)(Ph2P–N–PPh2)Fe(H)(κ1-Ph2P–NH–PPh2)2] (2), representing an example of cooperative bond activation with a homofunctional hemilabile ligand. This study demonstrates that hemilability of homofunctionalized ligands can be affected by careful adjustment of geometric parameters.
Co-reporter:Nicolas Frank, Katharina Hanau, Kimon Flosdorf and Robert Langer
Dalton Transactions 2013 vol. 42(Issue 31) pp:11252-11261
Publication Date(Web):11 Jun 2013
DOI:10.1039/C3DT51254A
A unique hydrido phosphine–borane iron(II) complex [(dppa)(Ph2P–N–P(BH3)Ph2)Fe(H)] (1) was obtained by the reaction of iron(II) chloride and two equivalents of bis(diphenylphosphino)amine (dppa) with an excess of sodium borohydride in acetonitrile–ethanol mixtures. Detailed investigations of the reaction revealed that a mixture of cis- and trans-[(dppa)2Fe(NCMe)2]2+ is formed prior to the reduction by sodium borohydride. Depending on the solvent, different products were obtained by the reduction: in acetonitrile–ethanol mixtures the hydrido phosphine–borane complex 1 is formed by formal insertion of BH3, while the reduction in pure acetonitrile results in the formation of the cationic complex trans-[(dppa)2Fe(H)(NCMe)](BH4) (4). Complex 4 is remarkably stable in ethanol and does not undergo phosphine–borane formation, even in the presence of excess sodium borohydride. This observation suggests that the phosphine–borane complex is generated by the reaction with the first equivalent of sodium borohydride with the participation of ethanol, followed by deprotonation or dihydrogen elimination. Experiments with similar diphosphine ligands, such as bis(diphenylphosphino)methane, did not yield a phosphine–borane complex, indicating the crucial role of the amine group in the observed reactivity.
Co-reporter:Nicolas Frank, Katharina Hanau, Kimon Flosdorf and Robert Langer
Dalton Transactions 2013 - vol. 42(Issue 31) pp:NaN11261-11261
Publication Date(Web):2013/06/11
DOI:10.1039/C3DT51254A
A unique hydrido phosphine–borane iron(II) complex [(dppa)(Ph2P–N–P(BH3)Ph2)Fe(H)] (1) was obtained by the reaction of iron(II) chloride and two equivalents of bis(diphenylphosphino)amine (dppa) with an excess of sodium borohydride in acetonitrile–ethanol mixtures. Detailed investigations of the reaction revealed that a mixture of cis- and trans-[(dppa)2Fe(NCMe)2]2+ is formed prior to the reduction by sodium borohydride. Depending on the solvent, different products were obtained by the reduction: in acetonitrile–ethanol mixtures the hydrido phosphine–borane complex 1 is formed by formal insertion of BH3, while the reduction in pure acetonitrile results in the formation of the cationic complex trans-[(dppa)2Fe(H)(NCMe)](BH4) (4). Complex 4 is remarkably stable in ethanol and does not undergo phosphine–borane formation, even in the presence of excess sodium borohydride. This observation suggests that the phosphine–borane complex is generated by the reaction with the first equivalent of sodium borohydride with the participation of ethanol, followed by deprotonation or dihydrogen elimination. Experiments with similar diphosphine ligands, such as bis(diphenylphosphino)methane, did not yield a phosphine–borane complex, indicating the crucial role of the amine group in the observed reactivity.
Co-reporter:W. Xu and R. Langer
Dalton Transactions 2015 - vol. 44(Issue 38) pp:NaN16790-16790
Publication Date(Web):2015/08/18
DOI:10.1039/C5DT02226F
A series of ruthenium(II) complexes with three novel PNX-type pincer ligands is reported, in which X denotes a heterocyclic donor group (PNX = Ph2PCH2CH2N(H)CH2-X, X = 2-pyridyl, 2-furanyl, 2-thiophenyl or 2-pyrrolyl). The reaction of [(Ph3P)3RuCl2] with one equivalent of ligand leads to the trans-dichloro complexes [(PNX)RuCl2(PPh3)] (1–4) for all ligands, whereas the complexation of [(Ph3P)3Ru(H)(Cl)(CO)] results in different types of complexes. The variation of the heterocyclic donor group is in line with different binding properties and a labilization of this group. Investigations on the catalytic activity of different types of ruthenium(II) complexes in hydrogenation and dehydrogenation reactions reveal that more labile bound heterocycle donors result in a decrease of catalytic productivity and activity.
Co-reporter:Maik Grätz, Andreas Bäcker, Lisa Vondung, Leon Maser, Arian Reincke and Robert Langer
Chemical Communications 2017 - vol. 53(Issue 53) pp:NaN7233-7233
Publication Date(Web):2017/04/27
DOI:10.1039/C7CC02335A
We report a novel method for the preparation of PBP-pincer complexes from bis(phosphine)boronium salts. The central (R3P)2HB-moiety in a palladium complex is demonstrated to be a L-type ligand, therewith completing a series of pincer-type complexes with Z-, X- and L-type boron-based ligands, respectively.



![1H-Imidazole, 2-[[bis(1-methylethyl)phosphino]methyl]-1-methyl-](http://img.cochemist.com/ccimg/230700/230621-88-2.png)
![1H-Imidazole, 2-[[bis(1-methylethyl)phosphino]methyl]-1-methyl-](http://img.cochemist.com/ccimg/230700/230621-88-2_b.png)
![Iron, (tetrahydrofuran)bis[1,1,1-trimethyl-N-(trimethylsilyl)silanaminato]-](/data/chemimg/113700/133984-10-8.png)
![Iron, (tetrahydrofuran)bis[1,1,1-trimethyl-N-(trimethylsilyl)silanaminato]-](/data/chemimg/113700/133984-10-8_b.png)
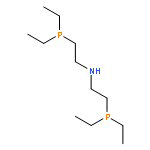
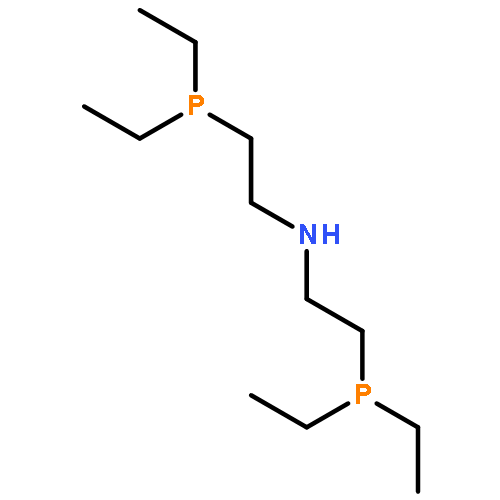


![Ethanamine, 2-(diphenylphosphino)-N-[2-(diphenylphosphino)ethyl]-](http://img.cochemist.com/ccimg/66600/66534-96-1.png)
![Ethanamine, 2-(diphenylphosphino)-N-[2-(diphenylphosphino)ethyl]-](http://img.cochemist.com/ccimg/66600/66534-96-1_b.png)
![Benzenamine, N,N-dimethyl-4-[(trimethylsilyl)thio]-](http://img.cochemist.com/ccimg/65300/65257-43-4.png)
![Benzenamine, N,N-dimethyl-4-[(trimethylsilyl)thio]-](http://img.cochemist.com/ccimg/65300/65257-43-4_b.png)
![[1,2-Bis(diphenyphosphino)ethane]dichloroiron(II)](http://img.cochemist.com/ccimg/41600/41536-18-9.png)
![[1,2-Bis(diphenyphosphino)ethane]dichloroiron(II)](http://img.cochemist.com/ccimg/41600/41536-18-9_b.png)




![[BIS(TRIMETHYLSILYL)]SELENIDE](http://img.cochemist.com/ccimg/4100/4099-46-1.png)
![[BIS(TRIMETHYLSILYL)]SELENIDE](http://img.cochemist.com/ccimg/4100/4099-46-1_b.png)

