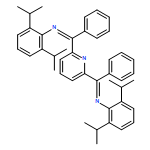
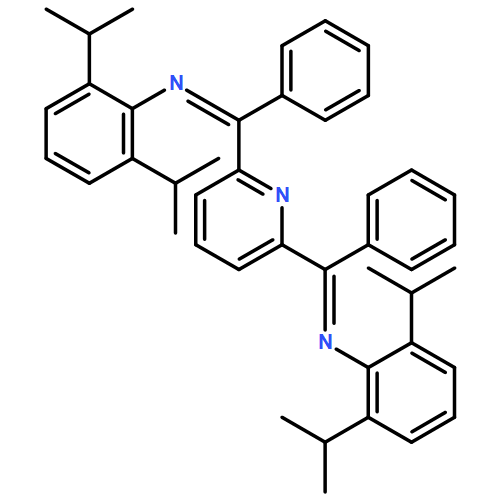
Co-reporter:Gyandshwar Kumar Rao;Mohammad Parsa Jamshidi;Jeremy I. G. Dawkins;Wendy Pell;Ilia Korobkov
Dalton Transactions 2017 vol. 46(Issue 20) pp:6518-6522
Publication Date(Web):2017/05/23
DOI:10.1039/C7DT01039G
A Mn(I) tris(2-pyridylmethyl)amine complex fac-[Mn(κ3-tpa) (CO)3]+OTf− carries out electrocatalytic hydrogen evolution from neutral water in acetonitrile. Bulk electrocatalytic studies showed that the catalyst functions with a moderate Faradaic efficiency and turn over frequency. DFT computations support the role of the tpa ligand as a shuttle to transfer of protons to the metal center.
Co-reporter:Gyandshwar Kumar Rao, Wendy Pell, Ilia Korobkov and Darrin Richeson
Chemical Communications 2016 vol. 52(Issue 51) pp:8010-8013
Publication Date(Web):31 May 2016
DOI:10.1039/C6CC03827A
New complexes, Mn{κ3-[2,6-{Ph2PNMe}2(NC5H3)]}(CO)3+Br− (1+Br−) and MnBr{κ2-(Ph2P)NMe(NC5H4)}(CO)3 (2), are reported and present new ligand environments for CO2 electrocatalytic reduction to CO. Compound 1+ presents a unique metal geometry for CO production (96%) in the absence of added water while 2 required addition of water and generated both CO and H2 products.
Co-reporter:Philip Bulsink, Ahlam Al-Ghamdi, Prajesh Joshi, Ilia Korobkov, Tom Woo and Darrin Richeson
Dalton Transactions 2016 vol. 45(Issue 21) pp:8885-8896
Publication Date(Web):28 Apr 2016
DOI:10.1039/C6DT00661B
The organometallic and coordination chemistry of rhenium(I) has been largely restricted to bidentate α-diimine ligation and facial tricarbonyl coordination geometries. The thermal transformation of bidentate bis(imino)pyridine and bidentate terpyridine complexes at 200–240 °C under nitrogen led to a family of Re(I) pincer complexes [κ3-2,6-{ArNCMe}2(NC5H3)]Re(CO)2X (ArC6H5, Me2C6H3, iPr2C6H3; X = Cl, Br) and (κ3-terpy)Re(CO)2X (X = Cl, Br). The synthesis, single crystal X-ray structural and spectroscopic characterization of these eight species documents their Re coordination geometries and demonstrates the accessibility of such compounds. The basic photophysical features of these compounds show significant elaboration in both number and intensity of the d–π* transitions observed in the UV-vis spectra relative to the bidentate starting materials and these spectra were analyzed using time-dependent DFT computations.
Co-reporter:Titel Jurca, Omar Ramadan, Ilia Korobkov, Darrin S. Richeson
Journal of Organometallic Chemistry 2016 Volume 802() pp:27-31
Publication Date(Web):15 January 2016
DOI:10.1016/j.jorganchem.2015.11.008
•Application of a bulky bis(imino)pyridine ligand, 2,6-iPr2C6H = N=CPh}2(NC5H3).•Bidentate coordination to yield fac-tricarbonylhalo Re(I) complexes.•Triflate complex obtained by reaction with AgOTf and opens potential reactive site on Re.Reported in this contribution are the synthesis and characterization of bidentate bis(imino)pyridine Re(I) complexes 2,6-{2,6-iPr2C6H3N = CPh}2(NC5H3)Re(CO)3X (X = Br (2), Cl (3), and OSO2CF3 (4)). Compounds 2 and 3 were prepared by direct reaction of bis(imino)pyridine ligand 2,6-{2,6-iPr2C6H3N = CPh}2(NC5H3) (1) with Re(CO)5X (X = Br, Cl) in toluene solution at 100 °C. Further treatment of 2 or 3 with Ag(OSO2CF3), in both instances, resulted in the formation of 4. Molecular structures of the new compounds (2, 3, and 4) were established by single crystal X-ray diffraction, and confirmed by elemental analysis and NMR studies.
Co-reporter:Gyandshwar Kumar Rao, Serge I. Gorelsky, Ilia Korobkov and Darrin Richeson
Dalton Transactions 2015 vol. 44(Issue 44) pp:19153-19162
Publication Date(Web):09 Oct 2015
DOI:10.1039/C5DT03515E
A series of monovalent group 11 complexes, [2,6-{Ph2PNMe}2(NC5H3)]CuBr 1, [2,6-{Ph2PNMe}2(NC5H3)]CuOTf 2, [2,6-{Ph2PNMe}2(NC5H3)]AgOTf 3, and [2,6-{Ph2PNMe}2(NC5H3)](AuCl)24, supported by a neutral PN3P ligand have been synthesized and characterized by multinuclear NMR and single crystal X-ray diffraction studies. The variation of the coordination properties were analyzed and electronic structure calculations have been carried out to provide insight on the bonding details in these complexes. The Cu(I) complexes displayed an unusual coordination geometry with a tridentate pincer ligand and an overall four coordinate trigonal pyramidal geometry. In contrast the Ag(I) analogue displayed a bidentate κ2-P,P′ ligation leaving the pyridyl-N atom uncoordinated and yielding a pyramidalized trigonal planar geometry around Ag. The bimetallic Au(I) complex completed the series and displayed a monodentate P-bonded ligand and a linear coordination geometry.
Co-reporter:Titel Jurca, Landon K. Hiscock, Ilia Korobkov, Christopher N. Rowley and Darrin S. Richeson
Dalton Transactions 2014 vol. 43(Issue 2) pp:690-697
Publication Date(Web):09 Oct 2013
DOI:10.1039/C3DT52227J
The autoionization reaction of neutral bis(imino)pyridine and SnX2 led to three compounds [{ArNCPh}2(NC5H3)]SnX+SnX3− (Ar = 2,6-(2,5-tBu2C6H3), X = Cl, Br; Ar = 2,6-(2,6-Me2C6H3), X = Cl) which display, within the same species, cations and anions possessing Sn(II) centers. Computational analysis compared the ligated Sn(II) cations with bis(imino)pyridine In(I) complexes that showed unprecedented weak metal–ligand covalent interactions, consistent with the In(I) 5s2 electrons remaining as an inert nonbonding pair. Analysis of the metal–ligand bonding indicates that the chloride ligand of the Sn(II) complex induces promotion of the metal 5s2 electron pair to a stereochemically active hybridized orbital, which, in turn, allows strong coordination of the bis(imino)pyridine to Sn.
Co-reporter:Titel Jurca, Ilia Korobkov, Serge I. Gorelsky, and Darrin S. Richeson
Inorganic Chemistry 2013 Volume 52(Issue 10) pp:5749-5756
Publication Date(Web):April 26, 2013
DOI:10.1021/ic302552v
The synthesis, characterization, and computational analysis of Tl(I) complexes bearing the bis(imino)pyridine scaffold, [{ArN═CPh}2(NC5H3)]Tl+(OTf)− (Ar = 2,6-Et2C6H33, 2,5-tBu2C6H3, 4), are reported. The cations of these species showed long Tl–N and Tl–OTf distances indicating only weak or no ligand coordination. Computational analysis of the interactions between the Tl cation and the ligands (orbital populations, bond order, and energy decomposition analysis) point to only minimal covalent interactions of the cation with the ligands. The weak ligand-to-metal donation allows for additional interactions between the Tl cation and arene rings that are either intramolecular, in the case of 3, or intermolecular. From benzene or toluene, 4 crystallizes with inverted sandwich structures having two [{(2,5-tBu2C6H3)N═CPh}2(NC5H3)]Tl+ cations bridged by either benzene or toluene. A density functional computational description of these Tl-arene contacts required exchange-correlation functionals with long-range exchange corrections (e.g., CAM-B3LYP or LC-PBE) and show that Tl-arene contacts are stabilized by noncovalent interactions.
Co-reporter:Dr. Titel Jurca;Dr. Wen-Ching Chen;Sheila Michel;Dr. Ilia Korobkov;Dr. Tiow-Gan Ong;Dr. Darrin S. Richeson
Chemistry - A European Journal 2013 Volume 19( Issue 13) pp:4278-4286
Publication Date(Web):
DOI:10.1002/chem.201203045
Abstract
The development of rhenium(I) chemistry has been restricted by the limited structural and electronic variability of the common pseudo-octahedral products fac-[ReX(CO)3L2] (L2=α-diimine). We address this constraint by first preparing the bidentate bis(imino)pyridine complexes [(2,6-{2,6-Me2C6H3NCPh}2C5H3N)Re(CO)3X] (X=Cl 2, Br 3), which were characterized by spectroscopic and X-ray crystallographic means, and then converting these species into tridentate pincer ligand compounds, [(2,6-{2,6-Me2C6H3NCPh}2C5H3N)Re(CO)2X] (X=Cl 4, Br 5). This transformation was performed in the solid-state by controlled heating of 2 or 3 above 200 °C in a tube furnace under a flow of nitrogen gas, giving excellent yields (≥95 %). Compounds 4 and 5 define a new coordination environment for rhenium(I) carbonyl chemistry where the metal center is supported by a planar, tridentate pincer-coordinated bis(imino)pyridine ligand. The basic photophysical features of these compounds show significant elaboration in both number and intensity of the d–π* transitions observed in the UV/Vis spec tra relative to the bidentate starting materials, and these spectra were analyzed using time-dependent DFT computations. The redox nature of the bis(imino)pyridine ligand in compounds 2 and 4 was examined by electrochemical analysis, which showed two ligand reduction events and demonstrated that the ligand reduction shifts to a more positive potential when going from bidentate 2 to tridentate 4 (+160 mV for the first reduction step and +90 mV for the second). These observations indicate an increase in electrostatic stabilization of the reduced ligand in the tridentate conformation. Elaboration on this synthetic methodology documented its generality through the preparation of the pseudo-octahedral rhenium(I) triflate complex [(2,6-{2,6-Me2C6H3NCPh}2C5H3N)Re(CO)2OTf] (7, 93 % yield).
Co-reporter:Titel Jurca, Sarah Ouanounou, Serge I. Gorelsky, Ilia Korobkov and Darrin S. Richeson
Dalton Transactions 2012 vol. 41(Issue 16) pp:4765-4771
Publication Date(Web):03 Feb 2012
DOI:10.1039/C2DT12112C
The bis(imino)pyridine scaffold provides support for the synthesis and characterization of unique Ag(I) pincer complexes [{ArNCPh}2(NPh)]Ag+(OTf)− (Ar = 2,5-tBu2C6H33; 2,6-iPr2C6H34). The bonding interactions between the cation–anion and between the bis(imino)pyridine ligand and the Ag centre are presented. Coordination of pyridine, toluene, 2-butyne and cyclooctene to the Ag centre led to the isolation and crystallographic characterization of labile transient adduct species. Bonding analysis of the adducts revealed conventional ligand–Ag coordination and important unconventional electron donation from the ligand to a π*-orbital of the bis(imino)pyridine group.
Co-reporter:Ian Mallov, Heather Spinney, Titel Jurca, Serge Gorelsky, Tara Burchell, Darrin Richeson
Inorganica Chimica Acta 2012 Volume 392() pp:5-9
Publication Date(Web):30 September 2012
DOI:10.1016/j.ica.2012.06.019
Deprotonated 2,2′-diindolylmethane functions as a dianionic chelating ligand for the isolation and characterization of the group 15 species {ArCH(3-MeC8H4N)2}PCl (2) and {ArCH(3-MeC8H4N)2}SbNMe2 (6, Ar = p-C6H4Br). The single crystal X-ray structure of 2 revealed a six-membered heterocycle featuring an N–P–N bonding motif and a terminal P–Cl function. The antimony complex 6 is a rare example of an N-heterocyclic aminostibine. DFT computations were used to examine the energies of isomers for these compounds and to describe the bonding features of the diindolyl fragment with the group 15 cation.Graphical abstractDeprotonated 2,2′-diindolylmethane dianion is a chelating ligand for the isolation and characterization of the group 15 species {3-Me(NC8H4)2CHAr}PCl and {3-Me(NC8H4)2CHAr}SbNMe2 (7, Ar = p-C6H4Br). DFT computations examine the energies and bonding features of these compounds.Highlights► First examples of group 15 (e.g. P and Sb) compounds of 2,2′-diindolylmethane dianion. ► Two different synthetic strategies employed in the synthesis. ► Antimony complex is a rare example of an N-heterocyclic aminostibine. ► Bonding features of the diindolyl fragment with the group 15 cation are examined through DFT computational analysis.
Co-reporter:Cory M. Widdifield, Titel Jurca, Darrin S. Richeson, David L. Bryce
Polyhedron 2012 Volume 35(Issue 1) pp:96-100
Publication Date(Web):16 March 2012
DOI:10.1016/j.poly.2012.01.003
While the synthetic utility and reactivity of the so-called “GaI” species is reasonably well established, its exact composition remains unknown. Using primarily multinuclear magnetic resonance techniques (and supported with powder X-ray diffraction measurements), we offer some new insights into the composition of “GaI”. Using 127I nuclear quadrupole resonance (NQR) experiments, it is clearly demonstrated that gallium diiodide (GaI2) is a significant component of this material, while GaI3 is not. This finding is in contrast with a prior literature account, which noted that Ga2I3 (i.e., one equivalent of GaI3 per equivalent of Ga metal) was present in significant amounts. Additional solid-state nuclear magnetic resonance (SSNMR) experiments using the 69/71Ga probe nuclei in multiple magnetic fields clearly establish, via its diagnostic Knight-shifted resonance, the presence of Ga0 metal, which is in agreement with prior literature. Taken together, a composition may be tentatively offered: that “GaI” is, to a large extent, two equivalents of Ga0 metal with two equivalents of GaI2, where the latter is composed of Ga+ and GaI4- ions. This composition may be represented as [Ga0]2[Ga]+[GaI4]−.Graphical abstractGallium-69/71 solid-state nuclear magnetic resonance and iodine-127 nuclear quadrupole resonance spectroscopies are used to provide insight into the structure and composition of Green’s synthetically useful “GaI” reagent.Highlights► 69/71Ga solid-state NMR and 127I NQR provide new details on “GaI” composition. ► Clear presence of GaI2 is detected for the first time. ► Prior reports which detect gallium metal are confirmed. ► “GaI” appears to be stable over long periods of time, if stored properly.
Co-reporter:Titel Jurca ; Ahmed Farghal ; Po-Heng Lin ; Ilia Korobkov ; Muralee Murugesu ;Darrin S. Richeson
Journal of the American Chemical Society 2011 Volume 133(Issue 40) pp:15814-15817
Publication Date(Web):September 7, 2011
DOI:10.1021/ja204562m
Bis(imino)pyridine pincer ligands in conjunction with two isothiocyanate ligands have been used to prepare two mononuclear Co(II) complexes. Both complexes have a distorted square-pyramidal geometry with the Co(II) centers lying above the basal plane. This leads to significant spin–orbit coupling for the d7 Co(II) ions and consequently to slow relaxation of the magnetization that is characteristic of Single-Molecule Magnet (SMM) behavior.
Co-reporter:Titel Jurca, Serge I. Gorelsky, Ilia Korobkov and Darrin S. Richeson
Dalton Transactions 2011 vol. 40(Issue 17) pp:4394-4396
Publication Date(Web):28 Mar 2011
DOI:10.1039/C1DT10411J
The bis(imino)pyridine scaffold provides for the synthesis and characterization of the unique Ag(I) pincer complexes [{ArNCPh}2(NPh)]Ag+(OTf)− (Ar = 2,5-tBu2C6H3; 2,6-iPr2C6H3). The similar covalent radii of Ag(I) and In(I), prompted a bonding comparison of these species with their In(I) analogues. Coordination of toluene to the Ag center revealed the stronger Lewis acidity of the metal site in these compounds relative to In(I) analogues.
Co-reporter:Titel Jurca, Ilia Korobkov, Glenn P. A. Yap, Serge I. Gorelsky, and Darrin S. Richeson
Inorganic Chemistry 2010 Volume 49(Issue 22) pp:10635-10641
Publication Date(Web):October 12, 2010
DOI:10.1021/ic1016438
The synthesis, characterization, and computational analysis of a series of low-valent, In(I) complexes bearing the bis(imino)pyridine scaffold, {Ar′N═CPh}2(NC5H3), is reported. A stepwise steric reduction of the aryl groups on the imine substituents around the coordination site, (Ar′ = 2,5-tBu2C6H3, 2,6-iPr2C6H3, 2,6-(CH3CH2)2C6H3) is explored through the spectroscopic and crystallographic examination of complexes [{Ar′N═CPh}2(NC5H3)]In+(OTf)− (1−3). Compounds 1−3 displayed long In−N and In−OTf distances indicating only weak or no coordination. Application of the ligand with Ar′ = 2,6-(CH3)2C6H3 led to an In(III) bis(imino)pyridine complex, [{2,6-Me2C6H3N═CPh}2(NC5H3)]In(OTf)2Cl 4 with coordinated ligand, chloride, and triflate groups. Computational analysis of the interactions between the In cation and the ligands (orbital populations, bond order, and energy decomposition analysis) point to only minimal covalent interactions of the In(I) cation with the ligands. Although it features three N donor centers, the bis(imino)pyridine ligand provides little ligand-to-metal donation. A thorough electronic structure analysis revealed a correlation of compound stability with the reduced contribution of the In(I) 5s lone electron pair to the highest occupied molecular orbital (HOMO) of the cation. This effect, originating from non-bonding orbital interactions between the metal and the ligand, is more prominent in sterically crowded environments. The discovery of this correlation may help in designing new low-valent complexes.
Co-reporter:Nathalie Lavoie, Serge I. Gorelsky, Zhixian Liu, Tara J. Burchell, Glenn P. A. Yap and Darrin S. Richeson
Inorganic Chemistry 2010 Volume 49(Issue 11) pp:5231-5240
Publication Date(Web):May 14, 2010
DOI:10.1021/ic1003963
Deprotonated N,N′-disubstituted 1,8-diaminonaphthalenes (R2DAN2−; R = (CH3)2CH, C6H5, 3,5-Me2C6H3) were incorporated into Ta(V) complexes employing two methods. The direct proton transfer reaction of the parent amine, 1,8-(RNH)2C10H6, with TaMe3Cl2 led to elimination of methane and formation of TaCl3[1,8-(RN)2C10H6] (1, 2). Reaction of the dilithiated amido species, Li2R2DAN, with TaMe3Cl2 or [Ta(NEt2)2Cl3 ] yielded TaMe3[1,8-(RN)2C10H6] (3, 4) and TaCl(NEt2)2[1,8-(RN)2C10H6] (5, 6), respectively. X-ray structural studies of these complexes revealed the flexible coordination behavior of R2DAN2− by demonstrating that the ligand bonded to Ta with a coordination array dependent on the identity of the other ligands bonded to tantalum. Computational analysis of these complexes confirmed that the energetic components for binding of R2DAN2− to these TaX32+ fragments were dominated by the electronic features of the metal fragment. Chemical transformations of the bound ligand were evaluated by reaction of compounds 5 and 6 with LiNMe2 and MeLi. Simple metathesis products Ta(NEt2)2NMe2[1,8-(iPrN)2C10H6] (R = iPr 7, R = 3,5-Me2(C6H3) 8) were obtained from reactions with LiNMe2. In contrast, when the R group of the DAN ligand was iPr, reaction with MeLi ultimately led to activation of the isopropyl group and formation of the metallaziridine [κ3-(Me2CN)(iPrN)C10H6]Ta(NEt2)2 (9) species via the elimination of methane.
Co-reporter:Titel Jurca, Karl Dawson, Ian Mallov, Tara Burchell, Glenn P. A. Yap and Darrin S. Richeson
Dalton Transactions 2010 vol. 39(Issue 5) pp:1266-1272
Publication Date(Web):24 Nov 2009
DOI:10.1039/B920047A
Attempted coordination of “GaII” with two new sterically bulky, aryl substituted bis(imino)pyridine ligands lead to GaIII species [2,6-{ArNCPh}2(NC5H3)]GaI2+GaI4− (Ar = 2,5-tBu2C6H3, 2,6-iPr2C6H3 = Dipp) arising from thermodynamically favorable disproportionation reactions. Examination of these reactions lead to isolation of a neutral radical species [2,6-{DippNCPh}2(NC5H3)]GaI2. Both EPR spectroscopy and DFT calculations on this compound indicate that the unpaired electron is localized in a di(imino)pyridine π* orbital of an anionic ligand with nearly zero contribution from the Ga or I centers. Reaction of {2,5-tBu2C6H3NCPh}2(NC5H3) with AlCl3 yielded an analogous Al(III) product, [{2,5-tBu2C6H3NCPh}2(NC5H3)]AlCl2+AlCl4−.
Co-reporter:Peter Dornan, Christopher N. Rowley, Jessica Priem, Seán T. Barry, Tara J. Burchell, Tom K. Woo and Darrin S. Richeson
Chemical Communications 2008 (Issue 31) pp:3645-3647
Publication Date(Web):20 Jun 2008
DOI:10.1039/B803732A
A novel method for the cyclotrimerization of dimethylcyanamide to form hexamethylmelamine has been developed using an aluminium amide catalyst; detailed DFT modelling of the catalytic cycle supports a triple insertion, nucleophilic ring closure, deinsertion mechanism.
Co-reporter:Christopher N. Rowley ; Tiow-Gan Ong ; Jessica Priem ; Tom K. Woo ;Darrin S. Richeson
Inorganic Chemistry 2008 Volume 47(Issue 20) pp:9660-9668
Publication Date(Web):September 24, 2008
DOI:10.1021/ic801028m
The synthesis of substituted guanidines is of significant interest for their use as versatile ligands and for the synthesis of bioactive molecules. Lithium amides supported by tetramethylethylenediamine have recently been shown to catalyze the guanylation of amines with carbodiimide. In this report, density functional theory (DFT) calculations are used to provide insight into the mechanism of this transformation. The mechanism identified through our calculations is a carbodiimide insertion into the lithium−amide bond to form a lithium guanidinate, followed by a proton transfer from the amine. The proton transfer transition state requires the dissociation of one of the chelating nitrogen centers of the lithium guanidinate, proton abstraction from the amine, and bond formation between the lithium center and the amine nitrogen. On the basis of this mechanism, further calculations predicted that aluminum amides would also function as active catalysts for the guanylation of amines. We confirm this experimentally and report the development of aluminum amides as a new main group catalyst for the guanylation of a range of electron-poor amines with carbodiimide.
Co-reporter:Christopher N. Rowley, Tiow-Gan Ong, Jessica Priem, Darrin S. Richeson and Tom K. Woo
Inorganic Chemistry 2008 Volume 47(Issue 24) pp:12024-12031
Publication Date(Web):November 12, 2008
DOI:10.1021/ic801739a
While lithium amides supported by tetramethylethylenediamine (TMEDA) are efficient catalysts in the synthesis of substituted guanidines via the guanylation of an amine with carbodiimide, as well as the guanylation of phosphines and conversion of alkynes into propiolamidines, aluminum amides are only efficient catalysts for the guanylation of amides. Density functional theory (DFT) calculations were used to explain this difference in activity. The origin of this behavior is apparent in the critical step where a proton is transferred from the substrate to a metal guanidinate. The activation energies of these steps are modest for amines, phosphines, and alkynes when a lithium catalyst was used, but are prohibitively high for the analogous reactions with phosphines and alkynes for aluminum amide catalysts. Energy decomposition analysis (EDA) indicates that these high activations energies are due to the high energetic cost of the detachment of a chelating guanidinate nitrogen from the aluminum in the proton transfer transition state. Amines are able to adopt an ideal geometry for facile proton transfer to the aluminum guanidinate and concomitant Al−N bond formation, while phosphines and alkynes are not.
Co-reporter:Heather A. Spinney, Ilia Korobkov and Darrin S. Richeson
Chemical Communications 2007 (Issue 16) pp:1647-1649
Publication Date(Web):12 Mar 2007
DOI:10.1039/B617434E
A 1,8-bis(alkylamido)naphthalene framework has been applied to the construction of N-heterocyclic arsenium and stibenium cations; a novel synthetic route, involving protonation of an ancillary amido ligand, was used to generate the base-stabilized stibenium cation.
Co-reporter:Nathalie Lavoie, Tiow-Gan Ong, Serge I. Gorelsky, Ilia Korobkov, Glenn P. A. Yap and Darrin S. Richeson
Organometallics 2007 Volume 26(Issue 26) pp:6586-6590
Publication Date(Web):November 23, 2007
DOI:10.1021/om700763v
Bis(imido)W(VI) complexes of dianionic N,N′-disubstituted 1,8-diamidonaphthalene (R2DAN2−) (R = iPr, 3,5-Me2C6H3) are reported, and an X-ray structure and computational analysis of W(═NtBu)2[1,8-(iPrN)2C10H6] 1 revealed that the nonplanar coordination of the iPr2DAN2− ligand to the W center is favored to allow for increased electron donation from the amido N centers to the W. Experimental studies revealed that the R2DAN2− amido linkage is the preferred reaction site with isocyanates to yield tridentate amido/N,N′-ureato ligands and that reactions with Al2Me6 lead to methylation of W and formation of heterobimetallic species via a μ2-bridging interaction of the R2DAN2− ligand.
Co-reporter:Gyandshwar Kumar Rao, Serge I. Gorelsky, Ilia Korobkov and Darrin Richeson
Dalton Transactions 2015 - vol. 44(Issue 44) pp:NaN19162-19162
Publication Date(Web):2015/10/09
DOI:10.1039/C5DT03515E
A series of monovalent group 11 complexes, [2,6-{Ph2PNMe}2(NC5H3)]CuBr 1, [2,6-{Ph2PNMe}2(NC5H3)]CuOTf 2, [2,6-{Ph2PNMe}2(NC5H3)]AgOTf 3, and [2,6-{Ph2PNMe}2(NC5H3)](AuCl)24, supported by a neutral PN3P ligand have been synthesized and characterized by multinuclear NMR and single crystal X-ray diffraction studies. The variation of the coordination properties were analyzed and electronic structure calculations have been carried out to provide insight on the bonding details in these complexes. The Cu(I) complexes displayed an unusual coordination geometry with a tridentate pincer ligand and an overall four coordinate trigonal pyramidal geometry. In contrast the Ag(I) analogue displayed a bidentate κ2-P,P′ ligation leaving the pyridyl-N atom uncoordinated and yielding a pyramidalized trigonal planar geometry around Ag. The bimetallic Au(I) complex completed the series and displayed a monodentate P-bonded ligand and a linear coordination geometry.
Co-reporter:Philip Bulsink, Ahlam Al-Ghamdi, Prajesh Joshi, Ilia Korobkov, Tom Woo and Darrin Richeson
Dalton Transactions 2016 - vol. 45(Issue 21) pp:NaN8896-8896
Publication Date(Web):2016/04/28
DOI:10.1039/C6DT00661B
The organometallic and coordination chemistry of rhenium(I) has been largely restricted to bidentate α-diimine ligation and facial tricarbonyl coordination geometries. The thermal transformation of bidentate bis(imino)pyridine and bidentate terpyridine complexes at 200–240 °C under nitrogen led to a family of Re(I) pincer complexes [κ3-2,6-{ArNCMe}2(NC5H3)]Re(CO)2X (ArC6H5, Me2C6H3, iPr2C6H3; X = Cl, Br) and (κ3-terpy)Re(CO)2X (X = Cl, Br). The synthesis, single crystal X-ray structural and spectroscopic characterization of these eight species documents their Re coordination geometries and demonstrates the accessibility of such compounds. The basic photophysical features of these compounds show significant elaboration in both number and intensity of the d–π* transitions observed in the UV-vis spectra relative to the bidentate starting materials and these spectra were analyzed using time-dependent DFT computations.
Co-reporter:Titel Jurca, Landon K. Hiscock, Ilia Korobkov, Christopher N. Rowley and Darrin S. Richeson
Dalton Transactions 2014 - vol. 43(Issue 2) pp:NaN697-697
Publication Date(Web):2013/10/09
DOI:10.1039/C3DT52227J
The autoionization reaction of neutral bis(imino)pyridine and SnX2 led to three compounds [{ArNCPh}2(NC5H3)]SnX+SnX3− (Ar = 2,6-(2,5-tBu2C6H3), X = Cl, Br; Ar = 2,6-(2,6-Me2C6H3), X = Cl) which display, within the same species, cations and anions possessing Sn(II) centers. Computational analysis compared the ligated Sn(II) cations with bis(imino)pyridine In(I) complexes that showed unprecedented weak metal–ligand covalent interactions, consistent with the In(I) 5s2 electrons remaining as an inert nonbonding pair. Analysis of the metal–ligand bonding indicates that the chloride ligand of the Sn(II) complex induces promotion of the metal 5s2 electron pair to a stereochemically active hybridized orbital, which, in turn, allows strong coordination of the bis(imino)pyridine to Sn.
Co-reporter:Heather A. Spinney, Ilia Korobkov and Darrin S. Richeson
Chemical Communications 2007(Issue 16) pp:NaN1649-1649
Publication Date(Web):2007/03/12
DOI:10.1039/B617434E
A 1,8-bis(alkylamido)naphthalene framework has been applied to the construction of N-heterocyclic arsenium and stibenium cations; a novel synthetic route, involving protonation of an ancillary amido ligand, was used to generate the base-stabilized stibenium cation.
Co-reporter:Peter Dornan, Christopher N. Rowley, Jessica Priem, Seán T. Barry, Tara J. Burchell, Tom K. Woo and Darrin S. Richeson
Chemical Communications 2008(Issue 31) pp:NaN3647-3647
Publication Date(Web):2008/06/20
DOI:10.1039/B803732A
A novel method for the cyclotrimerization of dimethylcyanamide to form hexamethylmelamine has been developed using an aluminium amide catalyst; detailed DFT modelling of the catalytic cycle supports a triple insertion, nucleophilic ring closure, deinsertion mechanism.
Co-reporter:Titel Jurca, Karl Dawson, Ian Mallov, Tara Burchell, Glenn P. A. Yap and Darrin S. Richeson
Dalton Transactions 2010 - vol. 39(Issue 5) pp:NaN1272-1272
Publication Date(Web):2009/11/24
DOI:10.1039/B920047A
Attempted coordination of “GaII” with two new sterically bulky, aryl substituted bis(imino)pyridine ligands lead to GaIII species [2,6-{ArNCPh}2(NC5H3)]GaI2+GaI4− (Ar = 2,5-tBu2C6H3, 2,6-iPr2C6H3 = Dipp) arising from thermodynamically favorable disproportionation reactions. Examination of these reactions lead to isolation of a neutral radical species [2,6-{DippNCPh}2(NC5H3)]GaI2. Both EPR spectroscopy and DFT calculations on this compound indicate that the unpaired electron is localized in a di(imino)pyridine π* orbital of an anionic ligand with nearly zero contribution from the Ga or I centers. Reaction of {2,5-tBu2C6H3NCPh}2(NC5H3) with AlCl3 yielded an analogous Al(III) product, [{2,5-tBu2C6H3NCPh}2(NC5H3)]AlCl2+AlCl4−.
Co-reporter:Gyandshwar Kumar Rao, Mohammad Parsa Jamshidi, Jeremy I. G. Dawkins, Wendy Pell, Ilia Korobkov and Darrin Richeson
Dalton Transactions 2017 - vol. 46(Issue 20) pp:NaN6522-6522
Publication Date(Web):2017/04/25
DOI:10.1039/C7DT01039G
A Mn(I) tris(2-pyridylmethyl)amine complex fac-[Mn(κ3-tpa) (CO)3]+OTf− carries out electrocatalytic hydrogen evolution from neutral water in acetonitrile. Bulk electrocatalytic studies showed that the catalyst functions with a moderate Faradaic efficiency and turn over frequency. DFT computations support the role of the tpa ligand as a shuttle to transfer of protons to the metal center.
Co-reporter:Gyandshwar Kumar Rao, Wendy Pell, Ilia Korobkov and Darrin Richeson
Chemical Communications 2016 - vol. 52(Issue 51) pp:NaN8013-8013
Publication Date(Web):2016/05/31
DOI:10.1039/C6CC03827A
New complexes, Mn{κ3-[2,6-{Ph2PNMe}2(NC5H3)]}(CO)3+Br− (1+Br−) and MnBr{κ2-(Ph2P)NMe(NC5H4)}(CO)3 (2), are reported and present new ligand environments for CO2 electrocatalytic reduction to CO. Compound 1+ presents a unique metal geometry for CO production (96%) in the absence of added water while 2 required addition of water and generated both CO and H2 products.
Co-reporter:Titel Jurca, Serge I. Gorelsky, Ilia Korobkov and Darrin S. Richeson
Dalton Transactions 2011 - vol. 40(Issue 17) pp:NaN4396-4396
Publication Date(Web):2011/03/28
DOI:10.1039/C1DT10411J
The bis(imino)pyridine scaffold provides for the synthesis and characterization of the unique Ag(I) pincer complexes [{ArNCPh}2(NPh)]Ag+(OTf)− (Ar = 2,5-tBu2C6H3; 2,6-iPr2C6H3). The similar covalent radii of Ag(I) and In(I), prompted a bonding comparison of these species with their In(I) analogues. Coordination of toluene to the Ag center revealed the stronger Lewis acidity of the metal site in these compounds relative to In(I) analogues.
Co-reporter:Titel Jurca, Sarah Ouanounou, Serge I. Gorelsky, Ilia Korobkov and Darrin S. Richeson
Dalton Transactions 2012 - vol. 41(Issue 16) pp:NaN4771-4771
Publication Date(Web):2012/02/03
DOI:10.1039/C2DT12112C
The bis(imino)pyridine scaffold provides support for the synthesis and characterization of unique Ag(I) pincer complexes [{ArNCPh}2(NPh)]Ag+(OTf)− (Ar = 2,5-tBu2C6H33; 2,6-iPr2C6H34). The bonding interactions between the cation–anion and between the bis(imino)pyridine ligand and the Ag centre are presented. Coordination of pyridine, toluene, 2-butyne and cyclooctene to the Ag centre led to the isolation and crystallographic characterization of labile transient adduct species. Bonding analysis of the adducts revealed conventional ligand–Ag coordination and important unconventional electron donation from the ligand to a π*-orbital of the bis(imino)pyridine group.

![2,6-Bis[1-(2,6-di-i-propylphenylimino)ethyl]pyridine](/data/chemimg/6300/204203-14-5.png)
![2,6-Bis[1-(2,6-di-i-propylphenylimino)ethyl]pyridine](/data/chemimg/6300/204203-14-5_b.png)