Co-reporter:Xiao Xia Han, Junbo Li, Ibrahim Halil Öner, Bing Zhao, Silke Leimkühler, Peter Hildebrandt, Inez M. Weidinger
Analytica Chimica Acta 2016 Volume 941() pp:35-40
Publication Date(Web):19 October 2016
DOI:10.1016/j.aca.2016.08.053
•Nickel electrodes were used without further functionalization as supports for various redox proteins.•It was possible to monitor the immobilized proteins via surface enhanced Raman spectroscopy.•The native structure of the immobilized proteins was preserved and they could exchange electrons with the Ni electrode.•The immobilized redox proteins worked as an electron relay between electrode and solubilized myoglobin.Practical use of many bioelectronic and bioanalytical devices is limited by the need of expensive materials and time consuming fabrication. Here we demonstrate the use of nickel electrodes as a simple and cheap solid support material for bioelectronic applications. The naturally nanostructured electrodes showed a surprisingly high electromagnetic surface enhancement upon light illumination such that immobilization and electron transfer reactions of the model redox proteins cytochrome b5 (Cyt b5) and cytochrome c (Cyt c) could be followed via surface enhanced resonance Raman spectroscopy. It could be shown that the nickel surface, when used as received, promotes a very efficient binding of the proteins upon preservation of their native structure. The immobilized redox proteins could efficiently exchange electrons with the electrode and could even act as an electron relay between the electrode and solubilized myoglobin. Our results open up new possibility for nickel electrodes as an exceptional good support for bioelectronic devices and biosensors on the one hand and for surface enhanced spectroscopic investigations on the other hand.
Co-reporter:Dr. Xiao Xia Han;Dr. Lei Chen;Dr. Uwe Kuhlmann;Claudia Schulz; Inez M. Weidinger; Peter Hildebrt
Angewandte Chemie International Edition 2014 Volume 53( Issue 9) pp:2481-2484
Publication Date(Web):
DOI:10.1002/anie.201310123
Abstract
Mesoporous M-TiO2 NCs, functionalized by PATP, can capture toxic anilines and phenols by azo coupling. Loading these nanodevices with Ag NPs offers the possibility for a sensitive quantitative determination of target compounds by SERRS spectroscopy, which allows multiplex detection because of the specific vibrational fingerprints. Sensitivity and selectivity can be further enhanced by concentrating the hybrid particles by an external magnet and compound-specific binding (anilines versus phenols). The bound toxic compounds can be degraded by TiO2-assisted photocatalysis after removal of the loaded hybrid particles from the sample solution with an external magnet. The degradation process can be enhanced in the presence of plasmonic Ag nanostructures.
Co-reporter:Dr. Xiao Xia Han;Dr. Lei Chen;Dr. Uwe Kuhlmann;Claudia Schulz; Inez M. Weidinger; Peter Hildebrt
Angewandte Chemie 2014 Volume 126( Issue 9) pp:2514-2517
Publication Date(Web):
DOI:10.1002/ange.201310123
Abstract
Benzidin und seine Derivate können im menschlichen Körper zu krebserregenden Aminen umgewandelt werden. Aus diesem Grund wird den potentiellen Risiken (z. B. Blasenkrebs) bei Kontakt mit kommerziell erhältlichen Benzidin-haltigen und analogen Farbstoffen wachsende Aufmerksamkeit geschenkt.1, 2 Einige Phenolderivate, die in der Herstellung alltäglicher Produkte eingesetzt werden, haben Ähnlichkeiten mit hormonaktiven Stoffen und erhöhen möglicherweise das Brustkrebsrisiko.3, 4 In der hier vorgestellten Arbeit haben wir zum ersten Mal multifunktionale Nanokomposite konstruiert, die den hochempfindlichen Nachweis, die Manipulation und lichtinduzierte Zersetzung toxischer Benzidin- und Phenolderivate ermöglichen. Magnetische TiO2-Nanokomposite (M-TiO2-NCs) wurden synthetisiert und mit p-Aminothiophenol (PATP) zur Bindung der Zielmoleküle über eine Azo-Kupplung funktionalisiert. Anhand der spezifischen spektroskopischen Signaturen in den oberflächenverstärkten Resonanz-Raman-Spektren (SERRS; surface-enhanced resonance Raman scattering) der Azoprodukte war es möglich, zielgerichtet toxische Aniline und Phenole zu identifizieren und schließlich deren Azoprodukte an der TiO2-Oberfläche photokatalytisch abzubauen, ein Prozess, der zudem durch Ausnutzung der Plasmonenresonanz eingebetteter Ag-Partikel verstärkt werden konnte.
Co-reporter:Jacek Kozuch, Iris von der Hocht, Florian Hilbers, Hartmut Michel, and Inez M. Weidinger
Biochemistry 2013 Volume 52(Issue 36) pp:
Publication Date(Web):August 5, 2013
DOI:10.1021/bi400535m
A novel oxo state of cytochrome c oxidase from Paracoccus denitrificans generated by successive addition of excess H2O2 and ammonia was investigated using resonance Raman (RR) spectroscopy. Addition of ammonia to the H2O2-generated artificial F state resulted in an upshift of the oxoferryl stretching vibration from 790 to 796 cm–1, indicating that ammonia influences ligation of the heme-bound oxygen in the binuclear center. Concomitantly performed RR measurements in the high-frequency region between 1300 and 1700 cm–1 showed a high-spin to low-spin transition of heme a3 upon generation of the F state that was not altered by addition of ammonia. Removal of H2O2 by addition of catalase resulted in the disappearance of the oxoferryl stretching vibration and major back transformation of heme a3 into the high-spin state. The ratio of high-spin to low-spin states was identical for intermediates created with and without ammonia, leading to the conclusion that ammonia does not interact directly with heme a3. Only for the ammonia-created state was a band at 612 nm observed in the UV–visible difference spectrum that was shifted to 608 nm after addition of catalase. Our results support the hypothesis by von der Hocht et al. [von der Hocht, I., et al. (2011) Proc. Natl. Acad. Sci. U.S.A. 108, 3964–3969] that addition of ammonia creates a novel oxo intermediate state called PN where ammonia binds to CuB once the oxo intermediate F state has been formed.
Co-reporter:Xiao Xia Han, Annette M. Schmidt, Gernot Marten, Anna Fischer, Inez M. Weidinger, and Peter Hildebrandt
ACS Nano 2013 Volume 7(Issue 4) pp:3212
Publication Date(Web):March 15, 2013
DOI:10.1021/nn305892j
Magnetic hybrid assemblies of Ag and Fe3O4 nanoparticles with biocompatibly immobilized myoglobin (Mb) were designed to detect and capture toxic targets (NO2–, CN–, and H2O2). Mb was covalently attached to chitosan-coated magnetic silver hybrid nanoparticles (M-Ag-C) via glutaraldehyde that serves as a linker for the amine groups of Mb and chitosan. As verified by surface-enhanced resonance Raman (SERR) spectroscopy, this immobilization strategy preserves the native structure of the bound Mb as well as the binding affinity for small molecules. On the basis of characteristic spectral markers, binding of NO2–, CN–, and H2O2 could be monitored and quantified, demonstrating the high sensitivity of this approach with detection limits of 1 nM for nitrite, 0.2 μM for cyanide, and 10 nM for H2O2. Owing to the magnetic properties, these particles were collected by an external magnet to achieve an efficient decontamination of the solutions as demonstrated by SERR spectroscopy. Thus, the present approach combines the highly sensitive analytical potential of SERR spectroscopy with an easy approach for decontamination of aqueous solutions with potential applications in food and in environmental and medical safety control.Keywords: decontamination; magnetic nanoparticles; myoglobin; SERR; silver nanoparticles; spectroscopic detection; toxic small molecule
Co-reporter:Arumugam Sivanesan, Khoa H. Ly, Witold Adamkiewicz, Konstanze Stiba, Silke Leimkühler, and Inez M. Weidinger
The Journal of Physical Chemistry C 2013 Volume 117(Issue 22) pp:11866-11872
Publication Date(Web):May 17, 2013
DOI:10.1021/jp4032578
Ag–TiO2 and Au–TiO2 hybrid electrodes were designed by covalent attachment of TiO2 nanoparticles to Ag or Au electrodes via an organic linker. The optical and electronic properties of these systems were investigated using the cytochrome b5 (Cyt b5) domain of sulfite oxidase, exclusively attached to the TiO2 surface, as a Raman marker and model redox enzyme. Very strong SERR signals of Cyt b5 were obtained for Ag-supported systems due to plasmonic field enhancement of Ag. Time-resolved surface-enhanced resonance Raman spectroscopic measurements yielded a remarkably fast electron transfer kinetic (k = 60 s–1) of Cyt b5 to Ag. A much lower Raman intensity was observed for Au-supported systems with undefined and slow redox behavior. We explain this phenomenon on the basis of the different potential of zero charge of the two metals that largely influence the electronic properties of the TiO2 island film.
Co-reporter:Arumugam Sivanesan, Govindasamy Kalaivani, Anna Fischer, Konstanze Stiba, Silke Leimkühler, and Inez M. Weidinger
Analytical Chemistry 2012 Volume 84(Issue 13) pp:5759
Publication Date(Web):May 31, 2012
DOI:10.1021/ac301001a
Silver nanoparticles with identical plasmonic properties but different surface functionalities are synthesized and tested as chemically selective surface-enhanced resonance Raman (SERR) amplifiers in a two-component protein solution. The surface plasmon resonances of the particles are tuned to 413 nm to match the molecular resonance of protein heme cofactors. Biocompatible functionalization of the nanoparticles with a thin film of chitosan yields selective SERR enhancement of the anionic protein cytochrome b5, whereas functionalization with SiO2 amplifies only the spectra of the cationic protein cytochrome c. As a result, subsequent addition of the two differently functionalized particles yields complementary information on the same mixed protein sample solution. Finally, the applicability of chitosan-coated Ag nanoparticles for protein separation was tested by in situ resonance Raman spectroscopy.
Co-reporter:Arumugam Sivanesan, Jacek Kozuch, H. Khoa Ly, Govindasamy Kalaivani, Anna Fischer and Inez M. Weidinger
RSC Advances 2012 vol. 2(Issue 3) pp:805-808
Publication Date(Web):05 Dec 2011
DOI:10.1039/C1RA00781E
Silica coated Ag nanoparticles with defined surface plasmon resonances are used to selectively detect and analyze protein cofactors in solution and on interfaces via surface enhanced resonance Raman spectroscopy. The silica coating has a surprisingly small effect on optical amplification but minimizes unwanted interactions between the protein and the nanoparticle.
Co-reporter:H. Khoa Ly, Christopher Köhler, Anna Fischer, Julia Kabuss, Felix Schlosser, Mario Schoth, Andreas Knorr, and Inez M. Weidinger
Langmuir 2012 Volume 28(Issue 13) pp:5819-5825
Publication Date(Web):March 8, 2012
DOI:10.1021/la205139g
Coral Pt islands films are deposited via electrochemical reduction on silica-coated nanostructured Ag electrodes. From these devices surface-enhanced (resonance) Raman [SE(R)R] signals of molecules exclusively attached to Pt are obtained with intensity up to 50% of the value determined for Ag. SE(R)R spectroscopic investigations are carried out with different probe molecules, silica-coating thicknesses, and excitation lines. Additionally, field enhancement calculations on Ag–SiO2–Pt support geometries are performed to elucidate the influence of the Pt island film nanostructure on the observed Raman intensities. It is concluded that the nonperfect coating of the Pt island film promotes the efficiency of the induced Pt SER activity. Comparison with similar measurements on Ag–SiO2–Au electrodes further suggests that the chemical nature of the deposited metal island film plays a minor role for the SE(R)R intensity.
Co-reporter:Arumugam Sivanesan, H. Khoa Ly, Jacek Kozuch, Murat Sezer, Uwe Kuhlmann, Anna Fischer and Inez M. Weidinger
Chemical Communications 2011 vol. 47(Issue 12) pp:3553-3555
Publication Date(Web):15 Feb 2011
DOI:10.1039/C0CC05058J
We present a preparation procedure for small sized biocompatibly coated Ag nanoparticles with tunable surface plasmon resonances. The conditions were optimised with respect to the resonance Raman signal enhancement of heme proteins and to the preservation of the native protein structure.
Co-reporter:Murat Sezer, Stefan Frielingsdorf, Diego Millo, Nina Heidary, Tillman Utesch, Maria-Andrea Mroginski, Bärbel Friedrich, Peter Hildebrandt, Ingo Zebger, and Inez M. Weidinger
The Journal of Physical Chemistry B 2011 Volume 115(Issue 34) pp:10368-10374
Publication Date(Web):July 15, 2011
DOI:10.1021/jp204665r
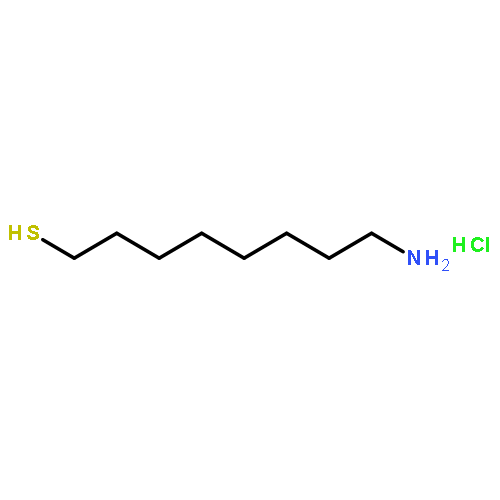
The role of the diheme cytochrome b (HoxZ) subunit in the electron transfer pathway of the membrane-bound [NiFe]-hydrogenase (MBH) heterotrimer from Ralstonia eutropha H16 has been investigated. The MBH in its native heterotrimeric state was immobilized on electrodes and subjected to spectroscopic and electrochemical analysis. Surface enhanced resonance Raman spectroscopy was used to monitor the redox and coordination state of the HoxZ heme cofactors while concomitant protein film voltammetric measurements gave insights into the catalytic response of the enzyme on the electrode. The entire MBH heterotrimer as well as its isolated HoxZ subunit were immobilized on silver electrodes coated with self-assembled monolayers of ω-functionalized alkylthiols, displaying the preservation of the native heme pocket structure and an electrical communication between HoxZ and the electrode. For the immobilized MBH heterotrimer, catalytic reduction of the HoxZ heme cofactors was observed upon H2 addition. The catalytic currents of MBH with and without the HoxZ subunit were measured and compared with the heterogeneous electron transfer rates of the isolated HoxZ. On the basis of the spectroscopic and electrochemical results, we conclude that the HoxZ subunit under these artificial conditions is not primarily involved in the electron transfer to the electrode but plays a crucial role in stabilizing the enzyme on the electrode.
Co-reporter:Jiu-Ju Feng;Ulrich Gernert;Peter Hildebrt
Advanced Functional Materials 2010 Volume 20( Issue 12) pp:1954-1961
Publication Date(Web):
DOI:10.1002/adfm.201000302
Abstract
A novel Ag–silica–Au hybrid device is developed that displays a long-range plasmon transfer of Ag to Au leading to enhanced Raman scattering of molecules largely separated from the optically excited Ag surface. A nanoscopically rough Ag surface is coated by a silica spacer of variable thickness from ∼1 to 21 nm and a thin Au film of ∼25 nm thickness. The outer Au surface is further functionalized by a self-assembled monolayer (SAM) for electrostatic binding of the heme protein cytochrome c (Cyt c) that serves as a Raman probe and model enzyme. High-quality surface-enhanced resonance Raman (SERR) spectra are obtained with 413 nm excitation, demonstrating that the enhancement results exclusively from excitation of Ag surface plasmons. The enhancement factor is estimated to be 2 × 104–8 × 103 for a separation of Cyt c from the Ag surface by 28–47 nm, corresponding to an attenuation of the enhancement by a factor of only 2–6 compared to Cyt c adsorbed directly on a SAM-coated Ag electrode. Upon immobilization of Cyt c on the functionalized Ag–silica–Au device, the native structure and redox properties are preserved as demonstrated by time- and potential-dependent SERR spectroscopy.
Co-reporter:Jiu-Ju Feng;Ulrich Gernert;Peter Hildebrt
Advanced Functional Materials 2010 Volume 20( Issue 12) pp:
Publication Date(Web):
DOI:10.1002/adfm.201090051
Co-reporter:Murat Sezer, Roberto Spricigo, Tillmann Utesch, Diego Millo, Silke Leimkuehler, Maria A. Mroginski, Ulla Wollenberger, Peter Hildebrandt and Inez M. Weidinger
Physical Chemistry Chemical Physics 2010 vol. 12(Issue 28) pp:7894-7903
Publication Date(Web):26 May 2010
DOI:10.1039/B927226G
Human sulfite oxidase (hSO) was immobilised on SAM-coated silver electrodes under preservation of the native heme pocket structure of the cytochrome b5 (Cyt b5) domain and the functionality of the enzyme. The redox properties and catalytic activity of the entire enzyme were studied by surface enhanced resonance Raman (SERR) spectroscopy and cyclic voltammetry (CV) and compared to the isolated heme domain when possible. It is shown that heterogeneous electron transfer and catalytic activity of hSO sensitively depend on the local environment of the enzyme. Increasing the ionic strength of the buffer solution leads to an increase of the heterogeneous electron transfer rate from 17 s−1 to 440 s−1 for hSO as determined by SERR spectroscopy. CV measurements demonstrate an increase of the apparent turnover rate for the immobilised hSO from 0.85 s−1 in 100 mM buffer to 5.26 s−1 in 750 mM buffer. We suggest that both effects originate from the increased mobility of the surface-bound enzyme with increasing ionic strength. In agreement with surface potential calculations we propose that at high ionic strength the enzyme is immobilised via the dimerisation domain to the SAM surface. The flexible loop region connecting the Moco and the Cyt b5 domain allows alternating contact with the Moco interaction site and the SAM surface, thereby promoting the sequential intramolecular and heterogeneous electron transfer from Moco via Cyt b5 to the electrode. At lower ionic strength, the contact time of the Cyt b5 domain with the SAM surface is longer, corresponding to a slower overall electron transfer process.
Co-reporter:Murat Sezer, Jiu-Ju Feng, H. Khoa Ly, Yanfei Shen, Takashi Nakanishi, Uwe Kuhlmann, Peter Hildebrandt, Helmuth Möhwald and Inez M. Weidinger
Physical Chemistry Chemical Physics 2010 vol. 12(Issue 33) pp:9822-9829
Publication Date(Web):11 Jun 2010
DOI:10.1039/C003082A
We have developed a new layered Au–Ag electrode for studying interfacial electron transfer processes by surface enhanced resonance Raman (SERR) spectroscopy. The device consists of a nanostructured Ag support which is separated from a Au film via a thin self-assembled monolayer (SAM) of amino-terminated mercaptanes (Cy-NH2, with y = 6, 8, 11). The Au film is biocompatibly coated to allow for binding of redox-active proteins. We have explored the performance of this device for analysing interfacial electron transfer processes by stationary and time-resolved SERR spectroscopy, using the heme protein cytochrome c (Cyt-c) as a benchmark protein. The SERRS intensity of Cyt-c on Ag-(Cy-NH2)-Au electrodes and Ag electrodes was comparable when the protein was electrostatically attached to the metal coated by a SAM of carboxyl-terminated mercaptanes (Cx-COOH) surface but 25 times higher upon covalent attachment via Cys102 to the bare Au surface. In the case of electrostatic adsorption the protein remained exclusively in its native state. Electron transfer between the protein and the Ag electrode occurred in an almost ideal Nernstian behaviour with a number of transferred electrons close to one (n = 0.8–0.9). Conversely, the covalent attached Cyt c showed two broad redox transitions (n = 0.3) and a partial conversion to a non-native species which remained redox inactive in the potential range from +0.1 to −0.3 V. For the electrostatically immobilised Cyt, apparent electron transfer rates of 0.8 and 49 s−1 were obtained for y = 11 and x = 15 and 10, respectively, indicating a fast long-distance electron transfer through the multilayer with the electron tunneling through the Cx-COOH SAM being the rate limiting step.
Co-reporter:Jiu-Ju Feng, Ulrich Gernert, Murat Sezer, Uwe Kuhlmann, Daniel H. Murgida, Christin David, Marten Richter, Andreas Knorr, Peter Hildebrandt and Inez M. Weidinger
Nano Letters 2009 Volume 9(Issue 1) pp:298-303
Publication Date(Web):December 22, 2008
DOI:10.1021/nl802934u
A nanostructured gold−silver-hybrid electrode for SER spectroelectrochemistry was developed which advantageously combines the electrochemical properties and chemical stability of Au and the strong surface enhancement of (resonance) Raman scattering by Ag. The layered device consists of a massive nanoscopically rough Ag electrode, a thin (2 nm) organic layer, and a ca. 20 nm thick Au film that may be coated by self-assembled monolayers for protein adsorption. The SERR-spectroscopic and electrochemical performance of this device is demonstrated using the heme protein cytochrome c as a benchmark model system, thereby extending, for the first time, SE(R)R studies of molecules on Au surfaces to excitation in the violet spectral range. The enhancement factor is only slightly lower than for Ag electrodes which can be rationalized in terms of an efficient transfer of plasmon resonance excitation from the Ag to the Au coating. This mechanism, which requires a thin dielectric layer between the two metals, is supported by theoretical calculations.
Co-reporter:Jiu-Ju Feng, Daniel H. Murgida, Uwe Kuhlmann, Tillmann Utesch, Maria Andrea Mroginski, Peter Hildebrandt and Inez M. Weidinger
The Journal of Physical Chemistry B 2008 Volume 112(Issue 47) pp:15202-15211
Publication Date(Web):October 31, 2008
DOI:10.1021/jp8062383
Iso-1 yeast cytochrome c (YCC) was adsorbed on Ag electrodes coated with self-assembled monolayers (SAMs) consisting either of 11-mercaptoundecanoic acid (MUA) or of 1:1 mixtures of MUA and either 11-mercaptoundecanol (MU) or 7-mercaptoheptanol (MH). The redox potentials and the apparent rate constants for the interfacial redox process as well as for the protein reorientation were determined by stationary surface-enhanced resonance Raman (SERR) and time-resolved SERR spectroscopy, respectively. For YCC immobilized on MUA and MUA/MU at pH 7.0 and 6.0, the negative shifts of the redox potentials with respect to that for the protein in solution can be rationalized in terms of the potential of the zero-charge determined by impedance measurements. The apparent electron transfer rate constants of YCC on MUA/MU and MU/MH at pH 6.0 were determined to be 8 and 18 s−1, respectively. A decrease of the relaxations constants by a factor of ca. 2 was found for pH 7.0, and a comparable low value was determined for a pure MUA even at pH 6.0. In each system, the rate constant for protein reorientation was found to be the same as that for the electron transfer, implying that protein reorientation is the rate limiting step for the interfacial redox process. This gating step is distinctly slower than that for horse heart cytochrome c (HHCC) observed previously under similar conditions (Murgida, D. H.; Hildebrandt, P. J. Am. Chem. Soc. 2001, 123, 4062−4068). The different rate constants of protein reorientation for both proteins and the variations of the rate constants for the different SAMs and pH are attributed to the electric field dependence of the free energy of activation which is assumed to be proportional to the product of the electric field strength and the molecular dipole moment of the protein. The latter quantity is determined by molecular dynamics simulations and electrostatic calculations to be more than 2 times larger for YCC than for HHCC. Moreover, the dipole moment vector and the heme plane constitute an angle of ca. 10 and 45° in YCC and HHCC, respectively. The different magnitudes and directions of the dipole moments as well as the different electric field strengths at the various SAM/protein interfaces allow for a qualitative description of the protein-, SAM-, and electrode-specific kinetics of the interfacial redox processes studied in this and previous works.
Co-reporter:Murat Sezer, Roberto Spricigo, Tillmann Utesch, Diego Millo, Silke Leimkuehler, Maria A. Mroginski, Ulla Wollenberger, Peter Hildebrandt and Inez M. Weidinger
Physical Chemistry Chemical Physics 2010 - vol. 12(Issue 28) pp:NaN7903-7903
Publication Date(Web):2010/05/26
DOI:10.1039/B927226G
Human sulfite oxidase (hSO) was immobilised on SAM-coated silver electrodes under preservation of the native heme pocket structure of the cytochrome b5 (Cyt b5) domain and the functionality of the enzyme. The redox properties and catalytic activity of the entire enzyme were studied by surface enhanced resonance Raman (SERR) spectroscopy and cyclic voltammetry (CV) and compared to the isolated heme domain when possible. It is shown that heterogeneous electron transfer and catalytic activity of hSO sensitively depend on the local environment of the enzyme. Increasing the ionic strength of the buffer solution leads to an increase of the heterogeneous electron transfer rate from 17 s−1 to 440 s−1 for hSO as determined by SERR spectroscopy. CV measurements demonstrate an increase of the apparent turnover rate for the immobilised hSO from 0.85 s−1 in 100 mM buffer to 5.26 s−1 in 750 mM buffer. We suggest that both effects originate from the increased mobility of the surface-bound enzyme with increasing ionic strength. In agreement with surface potential calculations we propose that at high ionic strength the enzyme is immobilised via the dimerisation domain to the SAM surface. The flexible loop region connecting the Moco and the Cyt b5 domain allows alternating contact with the Moco interaction site and the SAM surface, thereby promoting the sequential intramolecular and heterogeneous electron transfer from Moco via Cyt b5 to the electrode. At lower ionic strength, the contact time of the Cyt b5 domain with the SAM surface is longer, corresponding to a slower overall electron transfer process.
Co-reporter:Arumugam Sivanesan, H. Khoa Ly, Jacek Kozuch, Murat Sezer, Uwe Kuhlmann, Anna Fischer and Inez M. Weidinger
Chemical Communications 2011 - vol. 47(Issue 12) pp:NaN3555-3555
Publication Date(Web):2011/02/15
DOI:10.1039/C0CC05058J
We present a preparation procedure for small sized biocompatibly coated Ag nanoparticles with tunable surface plasmon resonances. The conditions were optimised with respect to the resonance Raman signal enhancement of heme proteins and to the preservation of the native protein structure.
Co-reporter:Murat Sezer, Jiu-Ju Feng, H. Khoa Ly, Yanfei Shen, Takashi Nakanishi, Uwe Kuhlmann, Peter Hildebrandt, Helmuth Möhwald and Inez M. Weidinger
Physical Chemistry Chemical Physics 2010 - vol. 12(Issue 33) pp:NaN9829-9829
Publication Date(Web):2010/06/11
DOI:10.1039/C003082A
We have developed a new layered Au–Ag electrode for studying interfacial electron transfer processes by surface enhanced resonance Raman (SERR) spectroscopy. The device consists of a nanostructured Ag support which is separated from a Au film via a thin self-assembled monolayer (SAM) of amino-terminated mercaptanes (Cy-NH2, with y = 6, 8, 11). The Au film is biocompatibly coated to allow for binding of redox-active proteins. We have explored the performance of this device for analysing interfacial electron transfer processes by stationary and time-resolved SERR spectroscopy, using the heme protein cytochrome c (Cyt-c) as a benchmark protein. The SERRS intensity of Cyt-c on Ag-(Cy-NH2)-Au electrodes and Ag electrodes was comparable when the protein was electrostatically attached to the metal coated by a SAM of carboxyl-terminated mercaptanes (Cx-COOH) surface but 25 times higher upon covalent attachment via Cys102 to the bare Au surface. In the case of electrostatic adsorption the protein remained exclusively in its native state. Electron transfer between the protein and the Ag electrode occurred in an almost ideal Nernstian behaviour with a number of transferred electrons close to one (n = 0.8–0.9). Conversely, the covalent attached Cyt c showed two broad redox transitions (n = 0.3) and a partial conversion to a non-native species which remained redox inactive in the potential range from +0.1 to −0.3 V. For the electrostatically immobilised Cyt, apparent electron transfer rates of 0.8 and 49 s−1 were obtained for y = 11 and x = 15 and 10, respectively, indicating a fast long-distance electron transfer through the multilayer with the electron tunneling through the Cx-COOH SAM being the rate limiting step.
















![Ferrate(2-), [7,12-diethenyl-3,8,13,17-tetramethyl-21H,23H-porphine-2,18-dipropanoato(4-)-κN21,κN22,κN23,κN24]-, hydrogen (1:2), (SP-4-2)-](/data/chemimg/522800/14875-96-8.png)
![Ferrate(2-), [7,12-diethenyl-3,8,13,17-tetramethyl-21H,23H-porphine-2,18-dipropanoato(4-)-κN21,κN22,κN23,κN24]-, hydrogen (1:2), (SP-4-2)-](/data/chemimg/522800/14875-96-8_b.png)

