Co-reporter:Shijun Wang, Shichen Yuan, Wei Chen, Qiming He, You-lee Hong, and Toshikazu Miyoshi
Macromolecules September 12, 2017 Volume 50(Issue 17) pp:6404-6404
Publication Date(Web):August 18, 2017
DOI:10.1021/acs.macromol.7b01462
The nucleation and growth mechanisms of semicrystalline polymers are a controversial topic in polymer science. In this work, we investigate the chain-folding pattern, packing structure, and crystal habits of poly(l-lactic acid) (PLLA) with a relatively low molecular weight, ⟨Mw⟩ = 46K g/mol, and PDI = 1.4 in single crystals formed from dilute amyl acetate (AA) solution (0.05 or 0.005 wt %) at a crystallization temperature (Tc) of 90, 50, or ∼0 °C. The crystal habits drastically changed from a facet lozenge shape at Tc = 90 °C to dendrites at ∼0 °C, whereas the chains adopt a thermodynamically stable α packing structure at both 90 and 0 °C. Comparing the experimental and simulated 13C–13C double quantum (DQ) buildup curves of 13C-labeled PLLA chains in crystals blended with nonlabeled chains at a mixing ratio of 1:9 indicates that the PLLA chains fold adjacently in multiple rows when the Tc ranges from 90 to ∼0 °C. The results at different length scales suggest that (i) a majority of the chains self-fold in dilute solution and form baby nuclei (intramolecular nucleation) and (ii) the intermolecular aggregation process (secondary nucleation), which is dominated by kinetics, results in morphological differences.
Co-reporter:Xiaoran Liu, Yuta Makita, You-lee Hong, Yusuke NishiyamaToshikazu Miyoshi
Macromolecules 2017 Volume 50(Issue 1) pp:
Publication Date(Web):December 23, 2016
DOI:10.1021/acs.macromol.6b02239
Inter- and intramolecular chemical reactions and their kinetics for 13C-labeled atactic-polyacrylonitrile (aPAN) powder heat-treated at 220–290 °C under air and vacuum were investigated by various solid-state nuclear magnetic resonance (ssNMR) techniques. By applying 13C direct polarization magic angle spinning (DPMAS) as well as through-bond and through-space double quantum/single quantum ssNMR techniques, it was concluded that aPAN heat-treated under air at 290 °C for 300 min adopted the ladder formation, namely, conjugated six-membered aromatic rings with partially cross-linked and oxidized rings and polyene components. In contrast, aPAN heat-treated under vacuum at the same condition thermally decomposed into oligomeric chains that were mainly composed of isolated aromatic rings connected by alkyl segments. Furthermore, early stages of the chemical reactions were investigated by 13C cross-polarization (CP) and DPMAS spectra. The latter provided quantitative information regarding the kinetics of the chemical reactions. As a result, it was shown that chemical reactions under oxygen occurred homogeneously with a higher activation energy (Ea) of 122 ± 3 kJ/mol compared to that of vacuum at 47 ± 2 kJ/mol. By comparing both chemical structures and kinetics under two different conditions, the chemical reaction mechanisms of aPAN will be discussed in detail.
Co-reporter:Jia Kang, Shichen Yuan, You-lee Hong, Wei Chen, Akihiro Kamimura, Akihiro Otsubo, and Toshikazu Miyoshi
ACS Macro Letters 2016 Volume 5(Issue 1) pp:65
Publication Date(Web):December 22, 2015
DOI:10.1021/acsmacrolett.5b00727
Despite numerous investigations on polymer processing, understanding the deformation mechanisms of semicrystalline polymer under uniaxial stretching is still challenging. In this work, 13C–13C Double Quantum (DQ) NMR was applied to trace the structural evolution of 13C-labeled isotactic polypropylene (iPP) chains inside the crystallites stretched to an engineering strain (e) of 21 at 100 °C. DQ NMR based on spatial proximity of 13C labeled nuclei proved conformational changes from the folded chains to the locally extended chains induced by stretching. By combining experimental findings with literature results on molecular dynamics, it was concluded that transportation of the crystalline chains plays a critical role to achieve large deformability of iPP.
Co-reporter:You-lee Hong, Wei Chen, Shichen Yuan, Jia Kang, and Toshikazu Miyoshi
ACS Macro Letters 2016 Volume 5(Issue 3) pp:355
Publication Date(Web):March 2, 2016
DOI:10.1021/acsmacrolett.6b00040
Over the last half century, a chain-folding structure of semicrystalline polymers has been debated in polymer science. Recently, 13C–13C double quantum (DQ) NMR spectroscopy combined with 13C selective isotope labeling has been developed to investigate re-entrance sites of the folded chains, mean values of adjacent re-entry number ⟨n⟩ and fraction ⟨F⟩ of semicrystalline polymers. This viewpoint highlights the versatile approaches of using solid-state (ss) NMR and isotope labeling for revealing (i) chain trajectory in melt- and solution-grown crystals, (ii) conformation of the folded chains in single crystals, (iii) self-folding in the early stage of crystallization, and (iv) unfolding of the folded chains under stretching.
Co-reporter:Wei Chen, Shijun Wang, Wei Zhang, Yutian Ke, You-lee Hong, and Toshikazu Miyoshi
ACS Macro Letters 2015 Volume 4(Issue 11) pp:1264
Publication Date(Web):October 30, 2015
DOI:10.1021/acsmacrolett.5b00685
Poly(l-lactide) (PLLA) and poly(d-lactide) (PDLA) alternatively pack with each other and form stereocomplex crystals (SCs). The crystal habits of SCs formed in the dilute solution highly depend on the molecular weight (⟨Mw⟩). In this study, we investigated chain-folding (CF) structure for 13C labeled PLLA (l-PLLA) chains in SCs with PDLAs that have either high or low ⟨Mw⟩s by employing an advanced Double Quantum (DQ) NMR. It was found that the ensemble average of the successive adjacent re-entry number ⟨n⟩ for the l-PLLA chains drastically change depending on ⟨Mw⟩s of the counter PDLA chains in the SCs. It was concluded that the CF structures of l-PLLA depending on ⟨Mw⟩s of PDLA determine the crystal habits of SCs.
Co-reporter:Shichen Yuan, Zhen Li, Jia Kang, You-lee Hong, Akihiro Kamimura, Akihiro Otsubo, and Toshikazu Miyoshi
ACS Macro Letters 2015 Volume 4(Issue 2) pp:143
Publication Date(Web):January 9, 2015
DOI:10.1021/mz5007969
Understanding the local packing structures of a disordered mesomorphic phase is a challenging issue in polymer characterization. In this work, 13C–13C through-space interactions, as well as a molecular dynamics analysis based on the reorientation of chemical shift anisotropy (CSA), were proposed for the evaluation of the local packing structure of the mesomorphic form of isotactic polypropylene (iPP). 13C–13C double quantum (DQ) buildup curves of 13C 15% CH3 selectively labeled iPP and spin-dynamics simulations demonstrated that the local packing structures in the mesomorphic form were very similar to the packing in the β phase. Moreover, centerband only detection of exchange (CODEX) NMR proved that the correlation time ⟨τc⟩ of the overall stem dynamics in the mesomorphic form followed the same Arrhenius line observed for the β phase, but it deviated from that for the α phase. Based on both structural and dynamic results, it was concluded that the local packing structure in the mesomorphic form was exceedingly close or the same as that of the β phase.
Co-reporter:Shichen Yuan, Zhen Li, You-lee Hong, Yutian Ke, Jia Kang, Akihiro Kamimura, Akihiro Otsubo, and Toshikazu Miyoshi
ACS Macro Letters 2015 Volume 4(Issue 12) pp:1382
Publication Date(Web):November 25, 2015
DOI:10.1021/acsmacrolett.5b00818
Understanding the structure formation of an ordered domain in the early stage of crystallization is one of the long-standing issues in polymer science. In this study, we investigate the chain trajectory of isotactic polypropylene (iPP) formed via rapid and deep quenching, using solid-state NMR spectroscopy. Comparisons of experimental and simulated 13C–13C double quantum (DQ) buildup curves demonstrated that individual iPP chains adopt adjacent re-entry sequences with an average folding number ⟨n⟩ = 3–4 in the mesomorphic form, assuming an adjacent re-entry fraction ⟨F⟩ of 100%. Therefore, long flexible polymer chains naturally fold in the early stage of crystallization, and folding-initiated nucleation results in formation of mesomorphic nanodomains.
Co-reporter:Zhen Li, You-lee Hong, Shichen Yuan, Jia Kang, Akihiro Kamimura, Akihiro Otsubo, and Toshikazu Miyoshi
Macromolecules 2015 Volume 48(Issue 16) pp:5752-5760
Publication Date(Web):August 5, 2015
DOI:10.1021/acs.macromol.5b01321
Chain-folding (CF) patterns, successive CF number ⟨n⟩ , and fraction ⟨F⟩ of 13C CH3 15% labeled isotactic polypropylene (iPP) in melt-grown crystals at two crystallization temperatures (Tcs) of 100 and 150 °C were investigated using 13C–13C Double quantum (DQ) NMR and spin-dynamics simulation. The former provided the α1 packing structure while the latter lead to the α2-rich sample with an α2 fraction of ca. 57%. It was demonstrated that the iPP chains adopt small cluster structures consisting of 6–8 stems connected via adjacent re-entry as mean structures in both α1 and α2 phases. There was no kinetic effect on ⟨F⟩ and ⟨n⟩ values, while kinetics did influence the folding directions of the chains under the assumptions of configurational and packing constraints. It was concluded that the CF process occurs at the growth front and dominates the chain-packing process of iPP α phases in a wide Tc.
Co-reporter:Xiaoran Liu, Wei Chen, You-lee Hong, Shichen Yuan, Shigeki Kuroki, and Toshikazu Miyoshi
Macromolecules 2015 Volume 48(Issue 15) pp:5300-5309
Publication Date(Web):July 27, 2015
DOI:10.1021/acs.macromol.5b01030
Solid-state (ss) NMR spectroscopy was applied to study the stabilization process of 30 wt % 13C-labeled atactic-polyacrylonitrile (a-PAN) heat-treated at various temperatures (Ts) under nitrogen and air. Direct polarization magic-angle spinning (DP/MAS) 13C NMR spectra provided quantitative information about the functional groups of stabilized a-PAN. Two dimensional (2D) refocused 13C–13C INADEQUATE and 1H–13C HETCOR NMR spectra gave through-bond and through-space correlations, respectively, of the complex intermediates and final structures of a-PAN stabilized at different Ts values. By comparing 1D and 2D NMR spectra, it was revealed that the stabilization process of a-PAN under nitrogen is initiated via cyclization, while the stabilization under air proceeds via dehydrogenation. Different initial processes lead to the isolated aromatic ring and ladder formation of the aromatic rings under nitrogen and air, respectively. Side reactions and intermediate structures are also discussed in detail. Through this work, the stabilization index (SI) was defined on the basis of the quantified C-1 and C-3 DP/MAS spectra. The former reached 0.87 at Ts = 370 °C, and further higher Ts values did not affect SI; however, the latter continuously increased up to 0.66 at Ts = 450 °C. All of the experimental results indicated that oxygen plays a vital role on the whole reaction process as well as the final products of stabilized a-PAN.
Co-reporter:You-lee Hong, Tadanori Koga, and Toshikazu Miyoshi
Macromolecules 2015 Volume 48(Issue 10) pp:3282-3293
Publication Date(Web):May 6, 2015
DOI:10.1021/acs.macromol.5b00079
The re-entrance sites, successive chain-folding number ⟨n⟩, and chain-folding fraction ⟨F⟩ of the chain-folding (CF) structure of 13C CH3-labeled isotactic poly(1-butene) (iPB1) with an weight-averaged molecular weight (⟨Mw⟩ = 37 K g/mol) in solution- and melt-grown crystals as a function of crystallization temperature (Tc) were determined using solid-state (SS) NMR. The solution- and melt-grown crystals possessed adjacent re-entry structures between the right- and left-handed stems along the (100) and (010) planes, which were invariant as a function of Tc. The adjacent re-entry structures in the former exhibited long-range order (⟨n⟩ ≥ 8) compared with that in the latter (⟨n⟩ ≥ 1.7–2). These results indicated that the concentration and entanglement of polymers play significant roles in the CF process and structural formation during the initial stage of crystallization, whereas kinetics does not. Transmission electron microscopy (TEM) revealed well-defined hexagonal and circular crystals grown from the solution state at Tc = 60 and ∼0 °C, respectively. The morphological and molecular-level structural data demonstrated that kinetics influences the structural formations of polymers differently at different length scales during crystallization. Moreover, SS-NMR, small-angle X-ray scattering (SAXS), and atomic force microscopy (AFM) indicated that the crystallinity (χc) and lamellar thickness (⟨lc⟩) of the melt-grown crystals are highly dependent on Tc, whereas in the solution-grown crystals, these parameters are independent of Tc. The experimental results and molecular dynamics, as reported in the literature, indicated that both χc and ⟨lc⟩ are primarily determined by the molecular dynamics of the stems after deposition of the chains on the growth front (late process).
Co-reporter:Wei Chen, Detlef Reichert, and Toshikazu Miyoshi
The Journal of Physical Chemistry B 2015 Volume 119(Issue 12) pp:4552-4563
Publication Date(Web):March 2, 2015
DOI:10.1021/acs.jpcb.5b00694
The molecular dynamics of Poly(l-lactic Acid) (PLLA) chains in the α phase was investigated by Solid-State NMR spectroscopy. 13C high-resolution NMR clearly indicates that the crystalline signals split into 2, 3, and 4 signals for the CH3, CH and CO groups, respectively at 25 °C, while the amorphous signals give a broad component at the bottom of the crystalline signals. 13C NMR spectra show that the crystalline line shape changes with increasing temperatures well above the glass transition temperature (Tg) and imply the presence of the molecular dynamics in the crystalline region. Comparisons of the evolution-time dependence of CODEX data and simulation results based on reorientation of chemical shift anisotropy (CSA) indicate that the chains in the α phase perform helical jump motions in the slow dynamic range at temperatures above 115 °C. The mixing-time dependence of the CODEX data yields an activation energy of Ea of (95 ± 8) kJ/mol for the helical jump motions. Moreover, two-dimensional exchange NMR with highly resolved signals for the CO group provides cross peaks among four well resolved signals due to the helical jumps. Comparison of 2D buildup curves of the cross peaks and calculated data determines that helical jump motions prefer largely uncorrelated random back-and-forth motions between the neighboring sites, possibly enabling large-scale chain diffusion in the crystalline regions.
Co-reporter:You-lee Hong and Toshikazu Miyoshi
ACS Macro Letters 2014 Volume 3(Issue 6) pp:556
Publication Date(Web):May 29, 2014
DOI:10.1021/mz500196s
Despite tremendous efforts over the last half-century to elucidate the chain-folding (CF) structure of semicrystalline polymers, the re-entrance sites of folded chains, the successive CF number n, and the adjacent re-entry fraction F have not been well characterized due to experimental limitations. In this report, 13C–13C double-quantum (DQ) NMR was used to determine for the first time the detailed CF structure of 13C CH3-labeled isotactic poly(1-butene) (iPB1) in solution-grown crystals blended with nonlabeled iPB1 across a wide range of crystallization temperatures (Tcs). Comparison of the results of DQ experiments and spin dynamics simulations demonstrated that the majority of individual chains possess completely adjacent re-entry structures at both Tc = 60 and ∼0 °C, as well as indicated that a low polymer concentration, not kinetics, leads to cluster formations of single molecules in dilute solution. The changes in crystal habits from hexagonal shapes at Tc = 60 °C to rounded shapes at ∼0 °C (kinetic roughness) are reasonably explained in terms of kinetically driven depositions of single molecule clusters on the growth front.
Co-reporter:Jia Kang and Toshikazu Miyoshi
Macromolecules 2014 Volume 47(Issue 9) pp:2993-3004
Publication Date(Web):April 16, 2014
DOI:10.1021/ma5004369
The chain packing, crystal thickness, molecular dynamics, and melting temperature of α-form isotactic polypropylene (iPP) drawn uniaxially at high temperatures of 100–150 °C were investigated using solid-state (SS) NMR and DSC. Two types of iPP samples with disordered (α1) and relatively ordered (α2-rich) packing structures were prepared via different thermal treatments and drawn up to an engineering strain (e) of approximately 20. High-resolution 13C NMR detected continuous α2 → α1 transformations in the original α2-rich samples over the entire deformation range at all drawing temperatures (Tds). A sudden α1 → α2 transformation was found to occur in the original α1 sample in the small e range of approximately 3–7 at Td = 140 °C. Then, in the late stage, the newly grown α2 structure reversely transformed into α1 structure with further increase in e, as observed in the original α2-rich sample. These results indicate that at least two different processes are involved in large deformations. On the basis of crystallographic constraints, the continuous α2 → α1 transformation over the entire deformation range is attributed to molecular-level melting and recrystallization facilitated by chain diffusion. The steep α1 → α2 transformation in the smaller e range is assigned to isotropic melting and recrystallization induced by stress. After the large deformations (e ≈ 20) of the original α2-rich and α1 samples at Td = 150 and 140 °C, respectively, 1H spin diffusion verified increases in the crystal thickness in both the former (14.1 at e = 0 → 20.1 nm at e = 20) and the latter (9.2 → 17.0 nm). Centerband-only detection of exchange (CODEX) NMR at 120 °C demonstrated that the correlation time (⟨τc⟩) of the helical jump for the former drastically decreased from ⟨τc⟩ = 52.4 ± 5.2 at e = 0 to 9.3 ± 1.8 ms at e = 20 but slightly increased from 4.2 ± 1.3 to 7.1 ± 0.9 ms for the latter. Additionally, DSC indicated that the melting temperature (Tm) for the former decreased considerably from 173 °C at e = 0 to 165 °C at e = 20, whereas the melting temperature (Tm) remained nearly invariant at 163 °C for the latter. On the basis of these findings, we conclude that the local packing structure plays a crucial role in determining the molecular dynamics of the stems and Tm of largely deformed iPP materials. The established relations among the structures, the dynamics, and the thermal properties provide a useful guide to achieving improved properties of iPP materials under processing.
Co-reporter:You-lee Hong and Toshikazu Miyoshi
ACS Macro Letters 2013 Volume 2(Issue 6) pp:501
Publication Date(Web):May 24, 2013
DOI:10.1021/mz300630j
A unique approach using 13C–13C double quantum (DQ) NMR combined with selective 13C isotope labeling is proposed to investigate the chain trajectory of the synthetic polymer in bulk crystals. Since the DQ buildup curve highly depends upon coupled spin number, topology, and internuclear distance, which originated from the chain trajectory of selectively 13C-labeled polymers, the adjacent re-entry site and fraction under finite chain-folding number can be determined.
Co-reporter:Zhen Li, Toshikazu Miyoshi, Mani K. Sen, Tadanori Koga, Akihiro Otsubo, and Akihiro Kamimura
Macromolecules 2013 Volume 46(Issue 16) pp:6507-6519
Publication Date(Web):August 13, 2013
DOI:10.1021/ma401032z
The order–disorder phenomenon and spatial heterogeneity of chain packing, partitions of stereodefects, and molecular dynamics of α form of isotactic polypropylene (iPP) samples, which are synthesized by Zieglar–Natta catalysts, are investigated by solid-state (SS) NMR. High-resolution 13C NMR under high-power TPPM decoupling at field strengths of 110 kHz allows observation of the order–disorder phenomenon in the chain-packing structures of α form. High isotacticity samples (isotacticity at pentad level, ⟨mmmm⟩ = 99.4%) give a maximum ordered packing (α2) fraction of 66% at crystallization temperature (Tc) of 155 °C while low stereoregularity samples (⟨mmmm⟩ = 91.0%) have only 47% at the same Tc. However, Mw (58.7–982 kg/mol) does not play a significant role in ordered packing formation. Using 13C-labeled CH3 of iPP, direct spatial correlations between the α2 and α1 structures are investigated by 13C detection of two-dimensional (2D) 1H–1H spin-diffusion (CHHC) experiments. The time dependence of the spin-diffusion polarization transferred signal intensities determines the average domain size of the α1 and α2 structures of iPP crystallized at 150 °C, which was found to be 40 nm under an assumption of 2D spin diffusion. Additionally, the 13C filter CPMAS NMR spectrum on 13C CH3-labeled iPP demonstrates that chemical defect is almost excluded from the crystalline region at Tc = 150 °C (defect free crystal) while ca. 2% is in melt quench sample. Moreover, 13C centerband-only detection of exchange experiments on α2-rich sample with highest ⟨mmmm⟩ = 99.4% indicate that crystalline dynamics follows a single Arrhenius plot with an activation energy of 116 kJ/mol across reported order–disorder transition temperatures (157–159 °C).
Co-reporter:Chuan Tang, Aoi Inomata, Yasuhiro Sakai, Hideaki Yokoyama, Toshikazu Miyoshi, and Kohzo Ito
Macromolecules 2013 Volume 46(Issue 17) pp:
Publication Date(Web):August 26, 2013
DOI:10.1021/ma401476g
The chemical modification of the pendant hydroxyl functional groups on cyclodextrins (CDs) significantly suppresses the hydrogen-bonding interactions between the cyclodextrin molecules and leads to the unique viscoelastic properties of hydroxypropylated polyrotaxane (HyPR) [Inomata et al. Macromolecules 2010, 43, 4660–4666]. HyPR consists of poly(ethylene glycol) (PEG) and α-CDs that are partially modified with a hydroxypropyl (Hy) group, setting them apart from other polyrotaxanes (PRs). The molecular dynamics of PR and HyPR with 25% (HyPR25) and 78% (HyPR78) modification ratios were investigated using various solid-state NMR techniques. Two-dimensional 1H–13C wide-line separation (WISE) NMR spectra of three samples demonstrated that the PEG chains provide two components of the restricted and the near-isotropic components in a fast motion limit at 329 K. The fraction of restricted dynamics of the threaded PEG chains was found to depend on the chemical modification ratio. In addition, WISE experiments proved that the CD side chains exhibit enhanced mobility when the modification fraction is increased. Centerband-only detection of exchange (CODEX) NMR was used to characterize the slow dynamics of both CD and PEG molecules with frequencies directly comparable to those used in viscoelastic measurements. The CD molecules undergo slow main-chain dynamics in HyPR78 in the mechanical-relaxation temperature range, whereas the other two systems do not. The temperature dependence of the correlation time ⟨τc⟩ determined by CODEX revealed Arrhenius behavior with a high activation energy (163 ± 16 kJ/mol), which is consistent with the previous viscoelastic result. The high activation energy for the dynamics of the CDs was interpreted in terms of cooperative motions with the threading PEG chains. The dependence of the evolution time of the CODEX data and simulation results indicated that the CD dynamics match random-jump and uniaxial rotation diffusion models. These results indicate that chemical modifications of the side groups can dramatically affect not only the molecular dynamics of both the CD main and side chains but also the threading of PEG chains across wide time scales.
Co-reporter:Wei Chen, Hao-Jan Sun, and Toshikazu Miyoshi
The Journal of Physical Chemistry B 2013 Volume 117(Issue 43) pp:13698-13709
Publication Date(Web):October 8, 2013
DOI:10.1021/jp4081492
An asymmetric tapered Janus bisamide supramolecule consisting of 1,4-bis[3,4,5-tris(alkan-1-yloxy)benzamido]benzene bisamide (abbreviated as C22PhBAEO3) can possess three-dimensional (3D) long-range order under mild thermal treatment conditions. To understand its structural formation and unique phase-transition processes, the locally detailed structure and molecular dynamics of its structural elements in disordered and ordered phases of C22PhBAEO3 were investigated using various solid-state (SS) NMR techniques at the atomic level. On the basis of the determined conformations and packing structures of the alkyl chains in ordered and disordered crystalline phases, along with the geometry and kinetic parameters of the structural elements’ dynamics, this study addresses the self-assembly, the phase-transition mechanisms, and the relationship between the structure and dynamics of these asymmetric Janus bisamide supramolecules.
Co-reporter:Toshikazu Miyoshi and Al Mamun
Polymer Journal 2012 44(1) pp:65-71
Publication Date(Web):August 10, 2011
DOI:10.1038/pj.2011.66
Isotactic-poly(1-butene) (iPB1) shows superior mechanical properties after crystal–crystal transitions. Recently, Miyoshi et al. found that crystalline stems in metastable tetragonal crystal perform uniaxial rotational diffusions accompanying side-chain conformational transitions in the fast motional limit (correlation time, τc <10−7 s; Macromolecules 2010, 43, 3986–3989.). In this study, molecular dynamics in stable trigonal crystal is investigated by solid-state nuclear magnetic resonance, which indicates that crystalline stems and side-chain conformations are completely fixed up to melting points (τc >10 s). In addition, lamellar thickness, l of iPB1 and a low isotacticity iPB1 (low_iPB1) with mmmm=78%, respectively, were investigated by small-angle X-ray scattering. The low_iPB1 sample shows very week supercooling dependence of l (~5 nm), whereas iPB1 shows strong supercooling dependence of l (10–28 nm). On the basis of molecular dynamics and l results, molecular dynamics effects on structures and unique mechanical property of iPB1 are discussed.










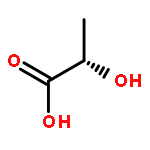
![POLY[OXY[(1R)-1-METHYL-2-OXO-1,2-ETHANEDIYL]]](http://img.cochemist.com/ccimg/27000/26917-25-9.png)
![POLY[OXY[(1R)-1-METHYL-2-OXO-1,2-ETHANEDIYL]]](http://img.cochemist.com/ccimg/27000/26917-25-9_b.png)
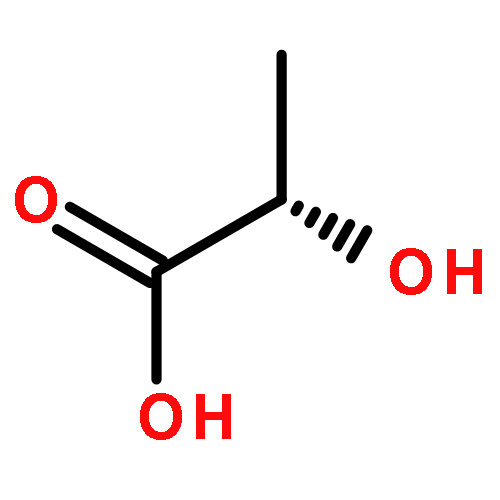


![Poly[oxy[(1S)-1-methyl-2-oxo-1,2-ethanediyl]]](http://img.cochemist.com/ccimg/26200/26161-42-2.png)
![Poly[oxy[(1S)-1-methyl-2-oxo-1,2-ethanediyl]]](http://img.cochemist.com/ccimg/26200/26161-42-2_b.png)