Co-reporter:Amanda J. Bischoff, Brandon M. Nelson, Zachary L. Niemeyer, Matthew S. Sigman, and Mohammad Movassaghi
Journal of the American Chemical Society November 1, 2017 Volume 139(Issue 43) pp:15539-15539
Publication Date(Web):October 4, 2017
DOI:10.1021/jacs.7b09541
The bis(pyridine)silver(I) permanganate promoted hydroxylation of diketopiperazines has served as a pivotal transformation in the synthesis of complex epipolythiodiketopiperazine alkaloids. This late-stage C–H oxidation chemistry is strategically critical to access N-acyl iminium ion intermediates necessary for nucleophilic thiolation of advanced diketopiperazines en route to potent epipolythiodiketopiperazine anticancer compounds. In this study, we develop an informative mathematical model using hydantoin derivatives as a training set of substrates by relating the relative rates of oxidation to various calculated molecular descriptors. The model prioritizes Hammett values and percent buried volume as key contributing factors in the hydantoin series while correctly predicting the experimentally observed oxidation sites in various complex diketopiperazine case studies. Thus, a method is presented by which to use simplified training molecules and resulting correlations to explain and predict reaction behavior for more complex substrates.
Co-reporter:Amanda J. Bischoff, Brandon M. Nelson, Zachary L. Niemeyer, Matthew S. Sigman, and Mohammad Movassaghi
Journal of the American Chemical Society November 1, 2017 Volume 139(Issue 43) pp:15539-15539
Publication Date(Web):October 4, 2017
DOI:10.1021/jacs.7b09541
The bis(pyridine)silver(I) permanganate promoted hydroxylation of diketopiperazines has served as a pivotal transformation in the synthesis of complex epipolythiodiketopiperazine alkaloids. This late-stage C–H oxidation chemistry is strategically critical to access N-acyl iminium ion intermediates necessary for nucleophilic thiolation of advanced diketopiperazines en route to potent epipolythiodiketopiperazine anticancer compounds. In this study, we develop an informative mathematical model using hydantoin derivatives as a training set of substrates by relating the relative rates of oxidation to various calculated molecular descriptors. The model prioritizes Hammett values and percent buried volume as key contributing factors in the hydantoin series while correctly predicting the experimentally observed oxidation sites in various complex diketopiperazine case studies. Thus, a method is presented by which to use simplified training molecules and resulting correlations to explain and predict reaction behavior for more complex substrates.
Co-reporter:Alyssa H. Antropow, Kun Xu, Rachel J. Buchsbaum, and Mohammad Movassaghi
The Journal of Organic Chemistry August 4, 2017 Volume 82(Issue 15) pp:7720-7720
Publication Date(Web):July 11, 2017
DOI:10.1021/acs.joc.7b01162
The synthesis of new agelastatin alkaloid derivatives and their anticancer evaluation in the context of the breast cancer microenvironment is described. A variety of N1-alkyl and C5-ether agelastatin derivatives were accessed via application of our strategy for convergent imidazolone synthesis from a common thioester along with appropriately substituted urea and alcohol components. These agelastatin derivatives were evaluated in our three-dimensional coculture assay for the effects of mammary fibroblasts on associated breast cancer cells. We have discovered that agelastatin alkaloids are potent modulators for cancer invasion and metastasis at noncytotoxic doses. Herein, we discuss the increased potency of (−)-agelastatin E as compared to (−)-agelastatin A in this capacity, in addition to identification of new agelastatin derivatives with activity that is statistically equivalent to (−)-agelastatin E. The chemistry described in this report provides a platform for the rapid synthesis of agelastatin derivatives with excellent potency (50–100 nM) as modulators for cancer invasion and metastasis.
Co-reporter:Dr. Taek Kang;Dr. Kolby L. White;Dr. Tyler J. Mann; Dr. Amir H. Hoveyda; Dr. Mohammad Movassaghi
Angewandte Chemie 2017 Volume 129(Issue 44) pp:14045-14048
Publication Date(Web):2017/10/23
DOI:10.1002/ange.201708088
AbstractThe first enantioselective total synthesis of (−)-deoxoapodine is described. Our synthesis of this hexacyclic aspidosperma alkaloid includes an efficient molybdenum-catalyzed enantioselective ring-closing metathesis reaction for the desymmetrization of an advanced intermediate that introduces the C5-quaternary stereocenter. After C21-oxygenation, the pentacyclic core was accessed by electrophilic C19-amide activation and transannular spirocyclization. A biogenetically inspired dehydrative C6-etherification reaction proved highly effective to secure the F-ring and the fourth contiguous stereocenter of (−)-deoxoapodine with complete stereochemical control.
Co-reporter:Kolby L. White and Mohammad Movassaghi
Journal of the American Chemical Society 2016 Volume 138(Issue 35) pp:11383-11389
Publication Date(Web):August 10, 2016
DOI:10.1021/jacs.6b07623
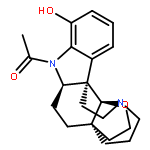
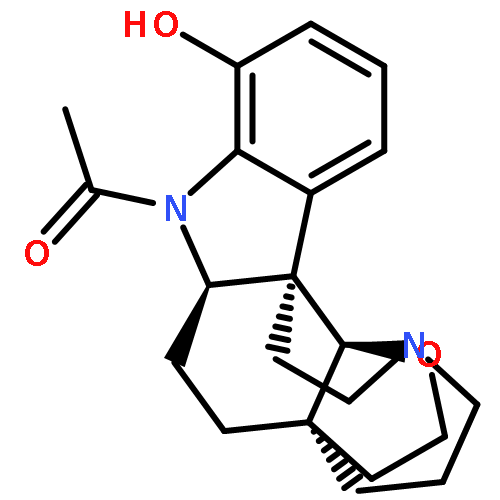
We report the first total syntheses of (+)-haplocidine and its N1-amide congener (+)-haplocine. Our concise synthesis of these alkaloids required the development of a late-stage and highly selective C–H oxidation of complex aspidosperma alkaloid derivatives. A versatile, amide-directed ortho-acetoxylation of indoline amides enabled our implementation of a unified strategy for late-stage diversification of hexacyclic C19-hemiaminal ether structures via oxidation of the corresponding pentacyclic C19-iminium ions. An electrophilic amide activation of a readily available C21-oxygenated lactam, followed by transannular cyclization and in situ trapping of a transiently formed C19-iminium ion, expediently provided access to hexacyclic C19-hemiaminal ether alkaloids (+)-fendleridine, (+)-acetylaspidoalbidine, and (+)-propionylaspidoalbidine. A highly effective enzymatic resolution of a non-β-branched primary alcohol (E = 22) allowed rapid preparation of both enantiomeric forms of a C21-oxygenated precursor for synthesis of these aspidosperma alkaloids. Our synthetic strategy provides succinct access to hexacyclic aspidosperma derivatives, including the antiproliferative alkaloid (+)-haplocidine.
Co-reporter:Stephen P. Lathrop; Matthew Pompeo; Wen-Tau T. Chang
Journal of the American Chemical Society 2016 Volume 138(Issue 24) pp:7763-7769
Publication Date(Web):May 31, 2016
DOI:10.1021/jacs.6b04072
The first biomimetic enantioselective total synthesis of (−)-communesin F based on a late-stage heterodimerization and aminal exchange is described. Our synthesis features the expedient diazene-directed assembly of two advanced fragments to secure the congested C3a–C3a′ linkage in three steps, followed by a highly efficient biogenetically inspired aminal reorganization to access the heptacyclic communesin core in only two additional steps. Enantioselective syntheses of the two fragments were developed, with highlights including the catalytic asymmetric halocyclization and diastereoselective oxyamination reactions of tryptamine derivatives, a stereoselective sulfinimine allylation, and an efficient cyclotryptamine–C3a-sulfamate synthesis by either a new silver-promoted nucleophilic amination or a rhodium-catalyzed C–H amination protocol. The versatile syntheses of the fragments, their stereocontrolled assembly, and the efficient aminal exchange as supported by in situ monitoring experiments, in addition to the final stage N1′-acylation of the communesin core, provide a highly convergent synthesis of (−)-communesin F.
Co-reporter:Richard P. Loach; Owen S. Fenton
Journal of the American Chemical Society 2016 Volume 138(Issue 3) pp:1057-1064
Publication Date(Web):January 4, 2016
DOI:10.1021/jacs.5b12392
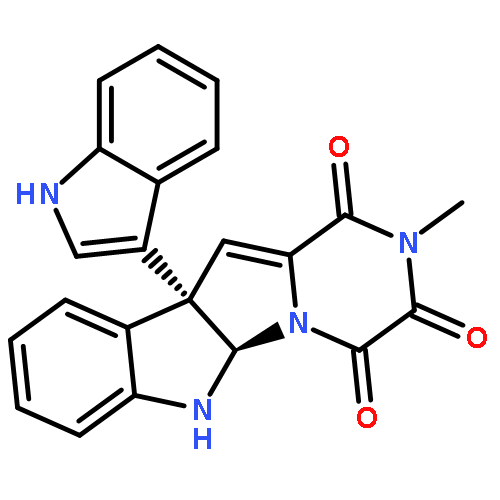
The concise, enantioselective total syntheses of (+)-asperazine (1), (+)-iso-pestalazine A (2), and (+)-pestalazine A (3) have been achieved by the development of a late-stage C3–C8′ Friedel–Crafts union of polycyclic diketopiperazines. Our modular strategy enables the union of complex polycyclic diketopiperazines in virtually their final forms, thus providing rapid and highly convergent assembly at the challenging quaternary stereocenter of these dimeric alkaloids. The significance of this carbon–carbon bond formation can be gauged by the manifold constraints that were efficiently overcome, namely the substantial steric crowding at both reactive sites, the nucleophilic addition of C8′ over N1′ to the C3 carbocation, and the multitude of reactivity posed by the use of complex diketopiperazine fragments in the coupling event. The success of the indoline π-nucleophile that evolved through our studies is notable given the paucity of competing reaction pathways observed in the presence of the highly reactive C3 carbocation generated. This first total synthesis of (+)-pestalazine A also allowed us to revise the molecular structure for this natural alkaloid.
Co-reporter:Justin Kim and Mohammad Movassaghi
Accounts of Chemical Research 2015 Volume 48(Issue 4) pp:1159
Publication Date(Web):April 6, 2015
DOI:10.1021/ar500454v
Natural products chemistry has historically been the prime arena for the discovery of new chemical transformations and the fountain of insights into key biological processes. It remains a fervent incubator of progress in the fields of chemistry and biology and an exchange mediating the flow of ideas between these allied fields of science. It is with this ethos that our group has taken an interest in and pursued the synthesis of a complex family of natural products termed the dimeric epipolythiodiketopiperazine (ETP) alkaloids. We present here an Account of the highly complex target molecules to which we pegged our ambitions, our systematic and relentless efforts toward those goals, the chemistry we developed in their pursuit, and the insight we have gained for their translational potential as potent anticancer molecules.The dimeric ETP alkaloids are fungal metabolites that feature a highly complex molecular architecture comprising a densely functionalized core structure with many stereogenic centers, six of which are fully substituted, and a pair of vicinal quaternary carbon stereocenters, decorated on polycyclic architectures in addition to the unique ETP motif that has been recognized as acid-, base-, and redox-sensitive. A cyclo-dipeptide consisting of an essential tryptophan residue and a highly variable ancillary amino acid lies at the core of these structures; investigation of the transformations that take this simplistic core to the complex alkaloids lies at the heart of our research program.The dimeric epidithiodiketopiperazine alkaloids have largely resisted synthesis on account of their complexity since the 1970s when the founding members of this class, chaetocin A (Hauser, D. et al. Helv. Chim. Acta 1970, 53, 1061) and verticillin A (Katagiri, K. et al. J. Antibiot. 1970, 23, 420), were first isolated. This was despite their potent cytotoxic and bacteriostatic activities, which were well appreciated at the time of their discovery. In the past decade, an increasing number of studies have uncovered powerful new biological processes that these molecules can uniquely effect, such as the inhibition of histone methyltransferases by chaetocin A (Greiner, D. et al. Nat. Chem. Biol. 2005, 1, 143). In fact, the complete collection of hexahydropyrroloindoline alkaloids features a diverse range of potent biological properties including cytotoxic, antitumor, antileukemic, antiviral, antibiotic, and antinematodal activities (Jiang, C.-S.; Guo, Y.-W. Mini-Rev. Med. Chem. 2011, 11, 728). This mélange of activities is reflective of their structural diversity.Under the precepts of retrobiosynthetic analysis, we have accomplished the syntheses of more than a dozen natural products, including members of the bionectin, calycanthaceous, chaetocin, gliocladin, naseseazine, and verticillin alkaloids. More importantly, these molecules have acted as venerable venues for the development of new strategies to address structural challenges including, but not limited to, C3–C3′ vicinal quaternary centers, heterodimeric linkages, C3–Csp2 linkages, diketopiperazine oxidation, stereoselective thiolation, homologue-specific polysulfidation, and C12-hydroxyl incorporation. Synthesis of these natural products has resulted in the structural confirmation, and sometimes revision such as the case of (+)-naseseazines A and B, as well as access to many plausible biogenetically relevant intermediates and new synthetic ETP derivatives. Furthermore, our studies have paved the way for the formulation of a comprehensive SAR profile and the identification of lead compounds with in vitro subnanomolar IC50’s against a broad range of cancer types.
Co-reporter:Timothy C. Adams, Joshua N. Payette, Jaime H. Cheah, and Mohammad Movassaghi
Organic Letters 2015 Volume 17(Issue 17) pp:4268-4271
Publication Date(Web):August 25, 2015
DOI:10.1021/acs.orglett.5b02059
The first total synthesis of (+)-luteoalbusins A and B is described. Highly regio- and diastereoselective chemical transformations in our syntheses include a Friedel–Crafts C3-indole addition to a cyclotryptophan-derived diketopiperazine, a late-stage diketopiperazine dihydroxylation, and a C11-sulfidation sequence, in addition to congener-specific polysulfane synthesis and cyclization to the corresponding epipolythiodiketopiperazine. We also report the cytoxicity of both alkaloids, and closely related derivatives, against A549, HeLa, HCT116, and MCF7 human cancer cell lines.
Co-reporter:Fan Liu, Mohammad Movassaghi
Tetrahedron Letters 2015 Volume 56(Issue 23) pp:2995-3000
Publication Date(Web):3 June 2015
DOI:10.1016/j.tetlet.2014.09.022
A series of tryptamine derived bisindole substrates were subjected to electrophilic activation of the functional grouping at their α-nitrogen in the form of iminium ions to enable cyclization onto the sterically hindered indole substructure. Our observations regarding divergent cyclization outcomes using electronically distinct bisindole substrates are described. Surprising preference for the Friedel–Crafts alkylation reaction and evidence for an intriguing reversible spirocyclization are discussed.
Co-reporter:Kolby L. White, Marius Mewald, and Mohammad Movassaghi
The Journal of Organic Chemistry 2015 Volume 80(Issue 15) pp:7403-7411
Publication Date(Web):July 12, 2015
DOI:10.1021/acs.joc.5b01023
The first mechanistic investigation of electrophilic amide activation of α,α-disubstituted tertiary lactams and the direct observation of key intermediates by in situ FTIR, 1H, 13C, and 19F NMR in our interrupted Bischler–Napieralski-based synthetic strategy to the aspidosperma alkaloids, including a complex tetracyclic diiminium ion, is discussed. The reactivity of a wide range of pyridines with trifluoromethanesulfonic anhydride was systematically examined, and characteristic IR absorption bands for the corresponding N-trifluoromethanesulfonylated pyridinium trifluoromethanesulfonates were assigned. The reversible formation of diiminium ether intermediates was studied, providing insight into divergent mechanistic pathways as a function of the steric environment of the amide substrate and stoichiometry of reagents. Importantly, when considering base additives during electrophilic amide activation, more hindered α-quaternary tertiary lactams require the use of non-nucleophilic pyridine additives in order to avoid deactivation via a competing desulfonylation reaction. The isolation and full characterization of a tetracyclic iminium trifluoromethanesulfonate provided additional correlation between in situ characterization of sensitive intermediates and isolable compounds involved in this synthetic transformation.
Co-reporter:Guoqiang Yang ; Petra Lindovska ; Dajian Zhu ; Justin Kim ; Peng Wang ; Ri-Yuan Tang ; Mohammad Movassaghi ;Jin-Quan Yu
Journal of the American Chemical Society 2014 Volume 136(Issue 30) pp:10807-10813
Publication Date(Web):July 9, 2014
DOI:10.1021/ja505737x
meta-C–H olefination, arylation, and acetoxylation of indolines have been developed using nitrile-containing templates. The combination of a monoprotected amino acid ligand and the nitrile template attached at the indolinyl nitrogen via a sulfonamide linkage is crucial for the meta-selective C–H functionalization of electron-rich indolines that are otherwise highly reactive toward electrophilic palladation at the para-positions. A wide range of synthetically important and advanced indoline analogues are selectively functionalized at the meta-positions.
Co-reporter:Stephen P. Lathrop and Mohammad Movassaghi
Chemical Science 2014 vol. 5(Issue 1) pp:333-340
Publication Date(Web):27 Sep 2013
DOI:10.1039/C3SC52451E
We describe the first application of our methodology for heterodimerization via diazene fragmentation towards the total synthesis of (−)-calycanthidine, meso-chimonanthine, and (+)-desmethyl-meso-chimonanthine. Our syntheses of these alkaloids feature an improved route to C3a-aminocyclotryptamines, an enhanced method for sulfamide synthesis and oxidation, in addition to a late-stage diversification leading to the first enantioselective total synthesis of (+)-desmethyl-meso-chimonanthine and its unambiguous stereochemical assignment. This versatile strategy for directed assembly of heterodimeric cyclotryptamine alkaloids has broad implications for the controlled synthesis of higher order derivatives with related substructures.
Co-reporter:Dr. Marius Mewald;Dr. Jonathan William Medley ;Dr. Mohammad Movassaghi
Angewandte Chemie 2014 Volume 126( Issue 43) pp:11818-11823
Publication Date(Web):
DOI:10.1002/ange.201405609
Abstract
Wir berichten über eine effiziente und hochstereoselektive Strategie zur Synthese von Aspidosperma-Alkaloiden, die auf der transannularen Cyclisierung einer chiralen Lactamvorstufe basiert. Aufgrund der bevorzugten Konformation der Cyclisierungsvorstufe werden in diesem Schlüsselschritt drei neue Stereozentren des resultierenden, vielseitigen, pentacyclischen Intermediats mit exzellenter Diastereoselektivität aufgebaut. Eine anschließende stereoselektive Epoxidierung mit nachfolgender Formamidreduktion unter milden Bedingungen ermöglichte die erste Totalsynthese der Aspidosperma-Alkaloide (−)-Mehranin und (+)-(6S,7S)-Dihydroxy-N-methylaspidospermidin. Eine abschließende, Scandiumtrifluormethansulfonat-vermittelte Dimerisierung von (−)-Mehranin führte zur ersten Totalsynthese von (−)-Methylenbismehranin.
Co-reporter:Dr. Marius Mewald;Dr. Jonathan William Medley ;Dr. Mohammad Movassaghi
Angewandte Chemie International Edition 2014 Volume 53( Issue 43) pp:11634-11639
Publication Date(Web):
DOI:10.1002/anie.201405609
Abstract
We report an efficient and highly stereoselective strategy for the synthesis of Aspidosperma alkaloids based on the transannular cyclization of a chiral lactam precursor. Three new stereocenters are formed in this key step with excellent diastereoselectivity due to the conformational bias of the cyclization precursor, leading to a versatile pentacyclic intermediate. A subsequent stereoselective epoxidation followed by a mild formamide reduction enabled the first total synthesis of the Aspidosperma alkaloids (−)-mehranine and (+)-(6S,7S)-dihydroxy-N-methylaspidospermidine. A late-stage dimerization of (−)-mehranine mediated by scandium trifluoromethanesulfonate completed the first total synthesis of (−)-methylenebismehranine.
Co-reporter:Richard P. Loach, Owen S. Fenton, Kazuma Amaike, Dustin S. Siegel, Erhan Ozkal, and Mohammad Movassaghi
The Journal of Organic Chemistry 2014 Volume 79(Issue 22) pp:11254-11263
Publication Date(Web):October 24, 2014
DOI:10.1021/jo502062z
A versatile strategy for C7-selective boronation of tryptophans, tryptamines, and 3-alkylindoles by way of a single-pot C2/C7-diboronation–C2-protodeboronation sequence is described. The combination of a mild iridium-catalyzed C2/C7-diboronation followed by an in situ palladium-catalyzed C2-protodeboronation allows efficient entry to valuable C7-boroindoles that enable further C7-derivatization. The versatility of the chemistry is highlighted by the gram-scale synthesis of C7-boronated N-Boc-L-tryptophan methyl ester and the rapid synthesis of C7-halo, C7-hydroxy, and C7-aryl tryptophan derivatives.
Co-reporter:Sunkyu Han, Karen C. Morrison, Paul J. Hergenrother, and Mohammad Movassaghi
The Journal of Organic Chemistry 2014 Volume 79(Issue 2) pp:473-486
Publication Date(Web):October 15, 2013
DOI:10.1021/jo4020358
A full account of our concise and enantioselective total syntheses of all known (−)-trigonoliimine alkaloids is described. Our retrobiosynthetic analysis of these natural products enabled identification of a single bistryptamine precursor as a precursor to all known trigonoliimines through a sequence of transformations involving asymmetric oxidation and reorganization. Our enantioselective syntheses of these alkaloids enabled the revision of the absolute stereochemistry of (−)-trigonoliimines A, B, and C. We report that trigonoliimines A, B, C and structurally related compounds showed weak anticancer activities against HeLa and U-937 cells.
Co-reporter:Alexis Coste, Justin Kim, Timothy C. Adams and Mohammad Movassaghi
Chemical Science 2013 vol. 4(Issue 8) pp:3191-3197
Publication Date(Web):29 May 2013
DOI:10.1039/C3SC51150B
The concise and efficient total synthesis of (+)-bionectins A and C is described. Our approach to these natural products features a new and scalable method for erythro-β-hydroxytryptophan amino acid synthesis, an intramolecular Friedel–Crafts reaction of a silyl-tethered indole, and a new mercaptan reagent for epipolythiodiketopiperazine (ETP) synthesis that can be unravelled under very mild conditions. In evaluating the impact of C12-hydroxylation, we have identified a unique need for an intramolecular variant of our Friedel–Crafts indolylation chemistry. Several key discoveries including the first example of permanganate-mediated stereoinvertive hydroxylation of the α-stereocenters of diketopiperazines as well as the first example of a direct triketopiperazine synthesis from a parent cyclo-dipeptide are discussed. Finally, the synthesis of (+)-bionectin A and its unambiguous structural assignment through X-ray analysis provides motivation for the reevaluation of its original characterization data and assignment.
Co-reporter:Nicolas Boyer, Karen C. Morrison, Justin Kim, Paul J. Hergenrother and Mohammad Movassaghi
Chemical Science 2013 vol. 4(Issue 4) pp:1646-1657
Publication Date(Web):24 Jan 2013
DOI:10.1039/C3SC50174D
The epipolythiodiketopiperazine (ETP) alkaloids are a highly complex class of natural products with potent anticancer activity. Herein, we report the application of a flexible and scalable synthesis, allowing the construction of dozens of ETP derivatives. The evaluation of these compounds against cancer cell lines in culture allows for the first expansive structure–activity relationship (SAR) to be defined for monomeric and dimeric ETP-containing natural products and their synthetic cognates. Many ETP derivatives demonstrate potent anticancer activity across a broad range of cancer cell lines and kill cancer cells via induction of apoptosis. Several traits that bode well for the translational potential of the ETP class of natural products include concise and efficient synthetic access, potent induction of apoptotic cell death, activity against a wide range of cancer types, and a broad tolerance for modifications at multiple sites that should facilitate small-molecule drug development, mechanistic studies, and evaluation in vivo.
Co-reporter:Jonathan William Medley and Mohammad Movassaghi
Chemical Communications 2013 vol. 49(Issue 92) pp:10775-10777
Publication Date(Web):27 Sep 2013
DOI:10.1039/C3CC44461A
The 1917 total synthesis of tropinone by Sir Robert Robinson represents a landmark achievement in organic synthesis. Decades ahead of its time in terms of its retrosynthetic logic and biomimetic approach, the elegant combination of these two elements in this synthesis continues to serve as an inspiration for the development of new and efficient strategies for complex molecule synthesis.
Co-reporter:Jonathan William Medley and Mohammad Movassaghi
Organic Letters 2013 Volume 15(Issue 14) pp:3614-3617
Publication Date(Web):July 5, 2013
DOI:10.1021/ol401465y
The development of a versatile method for the synthesis of spirocyclic pyrrolidinoindolines is discussed. Treatment of N-acyltryptamines with trifluoromethanesulfonic anhydride–2-chloropyridine reagent combination affords highly persistent spiroindoleninium ions that are subject to intra- and intermolecular addition at C2 by nucleophiles.
Co-reporter:Sunkyu Han, Dustin S. Siegel, Karen C. Morrison, Paul J. Hergenrother, and Mohammad Movassaghi
The Journal of Organic Chemistry 2013 Volume 78(Issue 23) pp:11970-11984
Publication Date(Web):October 23, 2013
DOI:10.1021/jo4020112
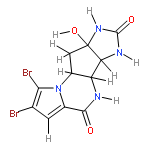
The full details of our enantioselective total syntheses of (−)-agelastatins A–F (1–6), the evolution of a new methodology for synthesis of substituted azaheterocycles, and the first side-by-side evaluation of all known (−)-agelastatin alkaloids against nine human cancer cell lines are described. Our concise synthesis of these alkaloids exploits the intrinsic chemistry of plausible biosynthetic precursors and capitalizes on a late-stage synthesis of the C-ring. The critical copper-mediated cross-coupling reaction was expanded to include guanidine-based systems, offering a versatile preparation of substituted imidazoles. The direct comparison of the anticancer activity of all naturally occurring (−)-agelastatins in addition to eight advanced synthetic intermediates enabled a systematic analysis of the structure–activity relationship within the natural series. Significantly, (−)-agelastatin A (1) is highly potent against six blood cancer cell lines (20–190 nM) without affecting normal red blood cells (>333 μM). (−)-Agelastatin A (1) and (−)-agelastatin D (4), the two most potent members of this family, induce dose-dependent apoptosis and arrest cells in the G2/M-phase of the cell cycle; however, using confocal microscopy, we have determined that neither alkaloid affects tubulin dynamics within cells.
Co-reporter:Nicolas Boyer and Mohammad Movassaghi
Chemical Science 2012 vol. 3(Issue 6) pp:1798-1803
Publication Date(Web):30 Mar 2012
DOI:10.1039/C2SC20270K
The first total synthesis of (+)-gliocladin B is described. Our concise and enantioselective synthesis takes advantage of a new regioselective Friedel–Crafts-based strategy to provide an efficient multigram-scale access to the C3-(3′-indolyl)hexahydropyrroloindole substructure, a molecular foundation present in a significant subset of epipolythiodiketopiperazine natural alkaloids. Our first-generation solution to (+)-gliocladin B involved the stereoselective formation of (+)-12-deoxybionectin A, a plausible biosynthetic precursor. Our synthesis clarified the C15 stereochemistry of (+)-gliocladin B and allowed its full structure confirmation. Further studies of a versatile dihydroxylated diketopiperazine provided a concise and efficient synthesis of (+)-gliocladin B as well as access to (+)-gliocladin C.
Co-reporter:Sunkyu Han, Dustin S. Siegel, Mohammad Movassaghi
Tetrahedron Letters 2012 Volume 53(Issue 29) pp:3722-3726
Publication Date(Web):18 July 2012
DOI:10.1016/j.tetlet.2012.04.121
The lithiation and electrophilic substitution of dimethyl triazones is described. Directed lithiation or tin–lithium exchange of dimethyl triazones afforded the corresponding dipole stabilized nucleophiles that were trapped with various electrophiles. Keto-triazone derivatives accessed by acylation of such nucleophiles were readily converted into the corresponding imidazolone heterocycles.The lithiation and electrophilic substitution of dimethyl triazones is described. Directed lithiation or tin–lithium exchange of dimethyl triazones afforded the corresponding dipole stabilized nucleophiles that were trapped with various electrophiles. Keto-triazone derivatives accessed by acylation of such nucleophiles were readily converted into the corresponding imidazolone heterocycles.
Co-reporter:Jonathan William Medley ;Dr. Mohammad Movassaghi
Angewandte Chemie International Edition 2012 Volume 51( Issue 19) pp:4572-4576
Publication Date(Web):
DOI:10.1002/anie.201200387
Co-reporter:Jonathan William Medley ;Dr. Mohammad Movassaghi
Angewandte Chemie 2012 Volume 124( Issue 19) pp:4650-4654
Publication Date(Web):
DOI:10.1002/ange.201200387
Co-reporter:Filip Kolundzic ; Mohammad N. Noshi ; Meiliana Tjandra ; Mohammad Movassaghi ;Scott J. Miller
Journal of the American Chemical Society 2011 Volume 133(Issue 23) pp:9104-9111
Publication Date(Web):May 3, 2011
DOI:10.1021/ja202706g
Catalytic enantioselective indole oxidation is a process of particular relevance to the chemistry of complex alkaloids, as it has been implicated in their biosynthesis. In the context of synthetic methodology, catalytic enantioselective indole oxidation allows a rapid and biomimetic entry into several classes of alkaloid natural products. Despite this potentially high utility in the total synthesis, reports of catalytic enantioselective indole oxidation remain sparse. Here we report a highly chemoselective catalytic system for the indole oxidation that delivers 3-hydroxy-indolenines with good chemical yields and moderate to high levels of enantio- and diastereoselectivity (up to 95:5 er and up to 92:8 dr). These results represent, to our knowledge, the most selective values yet reported in the literature for catalytic asymmetric indole oxidation. Furthermore, the utility of enantioenriched hydroxy-indolenines in stereospecific rearrangements is demonstrated.
Co-reporter:Sunkyu Han
Journal of the American Chemical Society 2011 Volume 133(Issue 28) pp:10768-10771
Publication Date(Web):June 13, 2011
DOI:10.1021/ja204597k
The concise and enantioselective total syntheses of (−)-trigonoliimines A, B, and C are described. Our unified strategy to all three natural products is based on asymmetric oxidation and reorganization of a single bistryptamine, a sequence of transformations with possible biogenetic relevance. We revise the absolute stereochemistry of (−)-trigonoliimines A, B, and C.
Co-reporter:Mohammad Movassaghi ; Omar K. Ahmad ;Stephen P. Lathrop
Journal of the American Chemical Society 2011 Volume 133(Issue 33) pp:13002-13005
Publication Date(Web):July 16, 2011
DOI:10.1021/ja2057852
A general strategy for the directed and stereocontrolled assembly of carbon–carbon linked heterodimeric hexahydropyrroloindoles is described. The stepwise union of complex amines in the form of mixed diazenes followed by photoexpulsion of dinitrogen in a solvent cage provides completely guided assembly at challenging Csp3–Csp3 and Csp3–Csp2 connections.
Co-reporter:Justin Kim
Journal of the American Chemical Society 2011 Volume 133(Issue 38) pp:14940-14943
Publication Date(Web):August 29, 2011
DOI:10.1021/ja206743v
Concise and enantioselective total syntheses of (+)-naseseazines A and B are described. Our regioselective and directed dimerization of diketopiperazines provides their critical C3–Csp2 linkages, an assembly with plausible biogenetic relevance. We revise the absolute stereochemistry of (+)-naseseazines A and B.
Co-reporter:Justin Kim
Journal of the American Chemical Society 2010 Volume 132(Issue 41) pp:14376-14378
Publication Date(Web):September 24, 2010
DOI:10.1021/ja106869s
A highly stereoselective and systematic strategy for the introduction of polysulfides in the synthesis of epipolythiodiketopiperazines is described. We report the first total synthesis of dimeric epitri- and epitetrathiodiketopiperazines.
Co-reporter:Mohammad Movassaghi, Dustin S. Siegel and Sunkyu Han
Chemical Science 2010 vol. 1(Issue 5) pp:561-566
Publication Date(Web):16 Aug 2010
DOI:10.1039/C0SC00351D
The pyrrole-imidazole family of marine alkaloids, derived from linear clathrodin-like precursors, constitutes a diverse array of structurally complex natural products. The bioactive agelastatins are members of this family that possess a tetracyclic molecular framework incorporating C4–C8 and C7–N12 bond connectivities. We provide a hypothesis for the formation of the unique agelastatin architecture that maximally exploits the intrinsic chemistry of plausible biosynthetic precursors. We report the concise enantioselective total syntheses of all known agelastatin alkaloids including the first total syntheses of agelastatins C, D, E, and F. Our gram-scale chemical synthesis of agelastatin A was inspired by our hypothesis for the biogenesis of the cyclopentane C-ring and required the development of new transformations including an imidazolone-forming annulation reaction and a carbohydroxylative trapping of imidazolones.
Co-reporter:Alison E. Ondrus, H. Ümit Kaniskan, Mohammad Movassaghi
Tetrahedron 2010 66(26) pp: 4784-4795
Publication Date(Web):
DOI:10.1016/j.tet.2010.04.006
Co-reporter:Justin Kim and Mohammad Movassaghi
Chemical Society Reviews 2009 vol. 38(Issue 11) pp:3035-3050
Publication Date(Web):23 Sep 2009
DOI:10.1039/B819925F
Since the synthesis of tropinone in the early 20th century, generations of chemists have looked to nature for inspiration in developing elegant syntheses of natural products. Analyzing molecules in their native context greatly enriches the chemist’s understanding of nature’s solution to the rapid generation of molecular complexity. In this tutorial review, we examine select examples from the alkaloid literature to highlight ways in which the close interplay of chemistry and biology have resulted in the development of elaborate biosynthetic hypotheses as well as strikingly beautiful syntheses. In addition, we present a case study on this topic through the lens of our total synthesis of (+)-11,11′-dideoxyverticillin A.
Co-reporter:Mohammad Movassaghi ; Meiliana Tjandra ;Jun Qi
Journal of the American Chemical Society 2009 Volume 131(Issue 28) pp:9648-9650
Publication Date(Web):June 25, 2009
DOI:10.1021/ja903790y
We describe the first total synthesis of (−)-himandrine, a member of the class II galbulimima alkaloids. Noteworthy features of this chemistry include a diastereoselective Diels−Alder reaction in the rapid synthesis of the tricycle ABC-ring system in an enantiomerically enriched form, the use of a formal [3+3] annulation strategy to secure the CDE-ring system with complete diastereoselection, and successful implementation of our biogenetically inspired oxidative spirocyclization of an advanced intermediate. The successful and direct late-stage formation of the F-ring in the hexacyclic core of himandrine drew on the power of biogenetic considerations and fully utilized the inherent chemistry of a plausible biosynthetic intermediate.
Co-reporter:Alison E. Ondrus and Mohammad Movassaghi
Chemical Communications 2009 (Issue 28) pp:4151-4165
Publication Date(Web):19 May 2009
DOI:10.1039/B903995N
The myrmicarins are a family of air- and temperature-sensitive alkaloids that possess unique structural features. Our concise enantioselective synthesis of the tricyclic myrmicarins enabled evaluation of a potentially biomimetic assembly of the complex members via direct dimerization of simpler structures. These studies revealed that myrmicarin 215B undergoes efficient and highly diastereoselective Brønsted acid-induced dimerization to generate a new heptacyclic structure, isomyrmicarin 430A. Mechanistic analysis demonstrated that heterodimerization between myrmicarin 215B and a conformationally restricted azafulvenium ion precursor afforded a functionalized isomyrmicarin 430A structure in a manner that was consistent with a highly efficient, non-concerted ionic process. Recent advancement in heterodimerization between tricyclic derivatives has enabled the preparation of strategically functionalized hexacyclic structures. The design and synthesis of structurally versatile dimeric compounds has greatly facilitated manipulation of these structures en route to more complex myrmicarin derivatives.
Co-reporter:Alison E. Ondrus and Mohammad Movassaghi
Organic Letters 2009 Volume 11(Issue 14) pp:2960-2963
Publication Date(Web):June 24, 2009
DOI:10.1021/ol9008552
Brønsted acid promoted reversible dimerization of myrmicarin 215B leads to formation of a new heptacyclic product, isomyrmicarin 430B, that possesses a C1,C2-trans,C2,C3-trans-substituted cyclopentane ring. Mechanistic studies illustrate that isomyrmicarin 430B arises by isomerization of isomyrmicarin 430A via fragmentation to tricyclic azafulvenium ions. Factors influencing the structure of heptacyclic isomyrmicarin products and potential relevance of this reversible vinyl pyrroloindolizine dimerization to the biosynthesis of complex myrmicarins are discussed.
Co-reporter:Justin Kim;James A. Ashenhurst
Science 2009 Volume 324(Issue 5924) pp:
Publication Date(Web):
DOI:10.1126/science.1170777
Abstract
The fungal metabolite (+)-11,11′-dideoxyverticillin A, a cytotoxic alkaloid isolated from a marine Penicillium sp., belongs to a fascinating family of densely functionalized, stereochemically complex, and intricate dimeric epidithiodiketopiperazine natural products. Although the dimeric epidithiodiketopiperazines have been known for nearly 4 decades, none has succumbed to total synthesis. We report a concise enantioselective total synthesis of (+)-11,11′-dideoxyverticillin A via a strategy inspired by our biosynthetic hypothesis for this alkaloid. Highly stereo- and chemoselective advanced-stage tetrahydroxylation and tetrathiolation reactions, as well as a mild strategy for the introduction of the epidithiodiketopiperazine core in the final step, were developed to address this highly sensitive substructure. Our rapid functionalization of the advanced molecular framework aims to mimic plausible biosynthetic steps and offers an effective strategy for the chemical synthesis of other members of this family of alkaloids.
Co-reporter:Mohammad Movassaghi, Grazia Piizzi, Dustin S. Siegel, Giovanni Piersanti
Tetrahedron Letters 2009 50(39) pp: 5489-5492
Publication Date(Web):
DOI:10.1016/j.tetlet.2009.07.102
Co-reporter:Dustin S. Siegel, Grazia Piizzi, Giovanni Piersanti and Mohammad Movassaghi
The Journal of Organic Chemistry 2009 Volume 74(Issue 24) pp:9292-9304
Publication Date(Web):November 25, 2009
DOI:10.1021/jo901926z
We report our full account of the enantioselective total synthesis of (−)-acylfulvene (1) and (−)-irofulven (2), which features metathesis reactions for the rapid assembly of the molecular framework of these antitumor agents. We discuss (1) the application of an Evans Cu-catalyzed aldol addition reaction using a strained cyclopropyl ketenethioacetal, (2) an efficient enyne ring-closing metathesis cascade reaction in a challenging setting, (3) the reagent IPNBSH for a late-stage reductive allylic transposition reaction, and (4) the final RCM/dehydrogenation sequence for the formation of (−)-acylfulvene (1) and (−)-irofulven (2).
Co-reporter:Mohammad Movassaghi Dr. ;OmarK. Ahmad
Angewandte Chemie 2008 Volume 120( Issue 46) pp:9041-9044
Publication Date(Web):
DOI:10.1002/ange.200802921
Co-reporter:Mohammad Movassaghi Dr.;MichaelA. Schmidt ;JamesA. Ashenhurst Dr.
Angewandte Chemie 2008 Volume 120( Issue 8) pp:1507-1509
Publication Date(Web):
DOI:10.1002/ange.200704960
Co-reporter:Mohammad Movassaghi Dr.;MichaelA. Schmidt ;JamesA. Ashenhurst Dr.
Angewandte Chemie International Edition 2008 Volume 47( Issue 8) pp:1485-1487
Publication Date(Web):
DOI:10.1002/anie.200704960
Co-reporter:Mohammad Movassaghi Dr. ;OmarK. Ahmad
Angewandte Chemie International Edition 2008 Volume 47( Issue 46) pp:8909-8912
Publication Date(Web):
DOI:10.1002/anie.200802921
Co-reporter:MatthewD. Hill Dr.
Chemistry - A European Journal 2008 Volume 14( Issue 23) pp:6836-6844
Publication Date(Web):
DOI:10.1002/chem.200800014
Abstract
Recent advances in pyrimidine synthesis are described. Modification of conventional strategies involving N-C-N fragment condensation with 1,3-dicarbonyl derivatives remains a common theme in current literature. Other methods, including N–C fragment condensation strategies, provide reactive intermediates capable of intramolecular cyclization and formation of pyrimidine derivatives. These recently developed methodologies offer a valuable addendum to azaheterocycle synthesis.
Co-reporter:
Nature Protocols 2007 2(8) pp:
Publication Date(Web):2007-08-16
DOI:10.1038/nprot.2007.280
Pyrimidines are found in a broad range of natural products and are contained in numerous pharmaceutically important molecules and functional materials1, 2, 3, 4. Many common strategies for synthesis of pyrimidine derivatives utilize condensation chemistry of amines and carbonyl-containing compounds1, 2, 3, 4, 5, 6, 7, 8, 9, 10. Limitations in some methods include lack of control in the positioning of substituents, necessarily large excess of at least one of the precursors, and often functional group incompatibility. Recent methods involving transition metal-catalyzed cross-coupling of heterocycles provide positional control in the introduction of substituents but require activated and functionalized heterocycles as substrates11, 12. We recently developed a convergent and single-step procedure for the preparation of pyrimidines from readily available substrates13. This new pyrimidine (3) annulation chemistry relies on the efficient condensation of N-vinyl or N-aryl secondary amides (1; see Fig. 1) with nitriles (2). The amide substrates are readily accessed via practical N-vinylation and N-arylation chemistry as well as a wide range of N-acylation protocols14, 15, 16, 17, 18. Many nitriles are commercially available and are rapidly prepared by a variety of synthetic methods, including the dehydration of primary amides19. The chemistry described here can provide fully substituted azaheterocycles with complete control in the position of the substituents.Recently, we reported a mild procedure for activation of secondary amides en route to pyridine derivatives employing 2-chloropyridine (2-ClPyr) as the acid scavenger in conjunction with trifluoromethanesulfonic anhydride (Tf2O) in dichloromethane (CH2Cl2)20, 21. In the context of contemporaneous mechanistic studies, we recognized that the activation of amides under these reaction conditions provides a uniquely electrophilic derivative subject to in situ nucleophilic trapping13, 22. Even weakly nucleophilic nitriles were found to add to these highly activated amide derivatives, leading to pyrimidines. As an example of this synthetic methodology, in the procedure described here, N-(3,4-dihydro-2H-pyran-5yl)benzamide (1a; Fig. 2) is activated with Tf2O in the presence of 2-ClPyr and trimethylacetonitrile (2a). In situ nitrile addition into the activated intermediate followed by cyclization and aromatization results in the synthesis of 4-tert-butyl-2-phenyl-7,8-dihydro-6Hpyrano[3,2-d]pyrimidine (3a; Fig. 2) in 86% (gram-scale).This protocol has been used to prepare a variety of azaheterocycles and fused bicycles13 (see Table 1). Importantly, this single-step procedure alleviates the need for isolation of activated intermediates. A wide range of aliphatic, aromatic and unsaturated amide and nitrile substrates were found compatible with this chemistry. N-vinyl amides were found to be significantly more reactive than N-aryl amides and required the presence of nitrile 2 during activation with Tf2O. These highly reactive substrates typically did not require heating above ambient temperature. Electron-rich and electron-deficient N-aryl amides gave the desired quinazoline derivatives while requiring heating above ambient temperature to promote the annulation step. The presence of nitrile upon activation of amide 1 was not mandatory in the case of these less reactive systems. The least reactive N-aryl substrates required heating to 140 °C in a microwave reactor to afford the desired quinazolines 3 in good yield. Where noted, it was beneficial to use a super-stoichiometric amount of nitrile to increase the yield of the desired product. A variety of functional groups were found to be compatible with this chemistry, and representative epimerizable amide and nitrile substrates were converted to the corresponding pyrimidine derivatives without loss of optical activity13.Steps 1–7: 10 minStep 8: 5 minStep 9: 7 minSteps 10 and 11: 10 minStep 12: 1 hStep 13–19: 35 minStep 20: 20 minSteps 21–24: 30 minStep 25: 20 minTroubleshooting advice can be found in Table 2.Yield, 86% (1.13 g); clear oil. TLC (20% vol/vol ethyl acetate in hexanes), Rf = 0.67 (UV, CAM). 1H NMR (500 MHz, CDCl3, 20 °C) δ: 8.42–8.37 (m, 2H, ArH), 7.48–7.38 (m, 3H, ArH), 4.27 (t, 2H, J = 5.1 Hz, CH2CH2CH2O), 2.98 (t, 2H, J = 6.7 Hz, CH2CH2CH2O), 2.20–2.14 (m, 2H, CH2CH2CH2O), 1.46 (s, 9H, C(CH3)3). 13C NMR (125 MHz, CDCl3, 20 °C) δ: 163.3, 154.8, 150.4, 147.6, 138.6, 129.4, 128.5, 127.7, 66.3, 38.2, 28.3, 28.1, 22.1. FTIR (neat, cm−1): 3,065 (w), 2,955 (m), 2,868 (w), 1,558 (m), 1,429 (m), 1,406 (s), 1,366 m), 1,353 (m). HRMS (ESI) (m/z) [M + H]+ calculated for C17H21N2O, 269.1654; found 269.1653. Analysis (calculated, found for C17H20N2O) C (76.09, 76.13), H (7.51, 7.52), N (10.44, 10.34).
Co-reporter:Mohammad Movassaghi Dr.;Michael A. Schmidt
Angewandte Chemie 2007 Volume 119(Issue 20) pp:
Publication Date(Web):5 APR 2007
DOI:10.1002/ange.200700705
Siamesische Zwillinge: Eine effiziente, konvergente Synthese der dimeren Hexahydropyrroloindol-Alkaloide (+)-Chimonanthin, (+)-Folicanthin und (−)-Calycanthin umfasst den Aufbau zweier vicinaler quartärer Stereozentren durch eine reduktive Dimerisierung. Auf diese Weise sind Gramm-Mengen des optisch aktiven Schlüsselintermediats zugänglich.
Co-reporter:Mohammad Movassaghi Dr.;Bin Chen Dr.
Angewandte Chemie 2007 Volume 119(Issue 4) pp:
Publication Date(Web):5 DEC 2006
DOI:10.1002/ange.200603302
Eine konvergente Synthese von tricyclischen Iminoalkoholen, die durch eine postulierte Biosynthese von Galbulimima-Alkaloiden inspiriert wurde, führt in einer sequenziellen α,α′-Alkylierung von unsymmetrischen Ketoiminen zum Aufbau von mindestens drei Stereozentren mit einem hohen Grad an Diastereoselektivität. Organokatalytische und asymmetrische Varianten dieser Methode ergänzen einen auf Organocuprat basierenden Ansatz (siehe Schema).
Co-reporter:Mohammad Movassaghi Dr.;Michael A. Schmidt
Angewandte Chemie International Edition 2007 Volume 46(Issue 20) pp:
Publication Date(Web):5 APR 2007
DOI:10.1002/anie.200700705
Conjoined twins: An efficient and convergent strategy for the synthesis of the dimeric hexahydropyrroloindole alkaloids, (+)-chimonanthine, (+)-folicanthine, and (−)-calycanthine, is described. The simultaneous formation of the vicinal quaternary stereocenters by using a reductive dimerization reaction provides an access to the optically active key intermediate on a gram scale.
Co-reporter:Mohammad Movassaghi Dr.;Bin Chen Dr.
Angewandte Chemie International Edition 2007 Volume 46(Issue 4) pp:
Publication Date(Web):5 DEC 2006
DOI:10.1002/anie.200603302
A convergent strategy for the synthesis of tricyclic imino alcohols was partly inspired by a postulated biosynthesis of galbulimima alkaloids. In this sequential α,α′ alkylation of unsymmetrical ketoimines, at least three stereocenters are created with a high level of diastereoselectivity. Organocatalytic and asymmetric variants of this methodology complement an organocuprate-based approach (see scheme).
Co-reporter:Mohammad Movassaghi Dr.;Grazia Piizzi Dr.;Dustin S. Siegel;Giovanni Piersanti Dr.
Angewandte Chemie International Edition 2006 Volume 45(Issue 35) pp:
Publication Date(Web):1 SEP 2006
DOI:10.1002/anie.200602011
Antitumor agents (−)-acylfulvene and (−)-irofulven are prepared in an approach that employs the powerful enyne ring-closing metathesis reaction to secure the spiro-bicyclic AB rings. Other key features of this synthesis include an efficient aldol-based introduction of the stereocenter at C2, a diazene-mediated reductive allylic transposition, and a ring-closing metathesis/oxidation sequence.
Co-reporter:Mohammad Movassaghi Dr.;Grazia Piizzi Dr.;Dustin S. Siegel;Giovanni Piersanti Dr.
Angewandte Chemie 2006 Volume 118(Issue 35) pp:
Publication Date(Web):1 SEP 2006
DOI:10.1002/ange.200602011
Tumortherapeutika: (−)-Acylfulven und (−)-Irofulven wurden mit einer Reaktionsfolge hergestellt, die die Enin-Ringschlussmetathese nutzt, um die spirobicyclischen AB-Ringe zu erhalten. Weitere Synthesemerkmale sind die effiziente Einführung des Stereozentrums an C2 durch eine Aldolreaktion, eine diazenvermittelte reduktive allylische Transposition und eine Ringschlussmetathese-Oxidations-Sequenz.
Co-reporter:Stephen P. Lathrop and Mohammad Movassaghi
Chemical Science (2010-Present) 2014 - vol. 5(Issue 1) pp:NaN340-340
Publication Date(Web):2013/09/27
DOI:10.1039/C3SC52451E
We describe the first application of our methodology for heterodimerization via diazene fragmentation towards the total synthesis of (−)-calycanthidine, meso-chimonanthine, and (+)-desmethyl-meso-chimonanthine. Our syntheses of these alkaloids feature an improved route to C3a-aminocyclotryptamines, an enhanced method for sulfamide synthesis and oxidation, in addition to a late-stage diversification leading to the first enantioselective total synthesis of (+)-desmethyl-meso-chimonanthine and its unambiguous stereochemical assignment. This versatile strategy for directed assembly of heterodimeric cyclotryptamine alkaloids has broad implications for the controlled synthesis of higher order derivatives with related substructures.
Co-reporter:Nicolas Boyer, Karen C. Morrison, Justin Kim, Paul J. Hergenrother and Mohammad Movassaghi
Chemical Science (2010-Present) 2013 - vol. 4(Issue 4) pp:NaN1657-1657
Publication Date(Web):2013/01/24
DOI:10.1039/C3SC50174D
The epipolythiodiketopiperazine (ETP) alkaloids are a highly complex class of natural products with potent anticancer activity. Herein, we report the application of a flexible and scalable synthesis, allowing the construction of dozens of ETP derivatives. The evaluation of these compounds against cancer cell lines in culture allows for the first expansive structure–activity relationship (SAR) to be defined for monomeric and dimeric ETP-containing natural products and their synthetic cognates. Many ETP derivatives demonstrate potent anticancer activity across a broad range of cancer cell lines and kill cancer cells via induction of apoptosis. Several traits that bode well for the translational potential of the ETP class of natural products include concise and efficient synthetic access, potent induction of apoptotic cell death, activity against a wide range of cancer types, and a broad tolerance for modifications at multiple sites that should facilitate small-molecule drug development, mechanistic studies, and evaluation in vivo.
Co-reporter:Justin Kim and Mohammad Movassaghi
Chemical Society Reviews 2009 - vol. 38(Issue 11) pp:NaN3050-3050
Publication Date(Web):2009/09/23
DOI:10.1039/B819925F
Since the synthesis of tropinone in the early 20th century, generations of chemists have looked to nature for inspiration in developing elegant syntheses of natural products. Analyzing molecules in their native context greatly enriches the chemist’s understanding of nature’s solution to the rapid generation of molecular complexity. In this tutorial review, we examine select examples from the alkaloid literature to highlight ways in which the close interplay of chemistry and biology have resulted in the development of elaborate biosynthetic hypotheses as well as strikingly beautiful syntheses. In addition, we present a case study on this topic through the lens of our total synthesis of (+)-11,11′-dideoxyverticillin A.
Co-reporter:Alexis Coste, Justin Kim, Timothy C. Adams and Mohammad Movassaghi
Chemical Science (2010-Present) 2013 - vol. 4(Issue 8) pp:NaN3197-3197
Publication Date(Web):2013/05/29
DOI:10.1039/C3SC51150B
The concise and efficient total synthesis of (+)-bionectins A and C is described. Our approach to these natural products features a new and scalable method for erythro-β-hydroxytryptophan amino acid synthesis, an intramolecular Friedel–Crafts reaction of a silyl-tethered indole, and a new mercaptan reagent for epipolythiodiketopiperazine (ETP) synthesis that can be unravelled under very mild conditions. In evaluating the impact of C12-hydroxylation, we have identified a unique need for an intramolecular variant of our Friedel–Crafts indolylation chemistry. Several key discoveries including the first example of permanganate-mediated stereoinvertive hydroxylation of the α-stereocenters of diketopiperazines as well as the first example of a direct triketopiperazine synthesis from a parent cyclo-dipeptide are discussed. Finally, the synthesis of (+)-bionectin A and its unambiguous structural assignment through X-ray analysis provides motivation for the reevaluation of its original characterization data and assignment.
Co-reporter:Mohammad Movassaghi, Dustin S. Siegel and Sunkyu Han
Chemical Science (2010-Present) 2010 - vol. 1(Issue 5) pp:NaN566-566
Publication Date(Web):2010/08/16
DOI:10.1039/C0SC00351D
The pyrrole-imidazole family of marine alkaloids, derived from linear clathrodin-like precursors, constitutes a diverse array of structurally complex natural products. The bioactive agelastatins are members of this family that possess a tetracyclic molecular framework incorporating C4–C8 and C7–N12 bond connectivities. We provide a hypothesis for the formation of the unique agelastatin architecture that maximally exploits the intrinsic chemistry of plausible biosynthetic precursors. We report the concise enantioselective total syntheses of all known agelastatin alkaloids including the first total syntheses of agelastatins C, D, E, and F. Our gram-scale chemical synthesis of agelastatin A was inspired by our hypothesis for the biogenesis of the cyclopentane C-ring and required the development of new transformations including an imidazolone-forming annulation reaction and a carbohydroxylative trapping of imidazolones.
Co-reporter:Nicolas Boyer and Mohammad Movassaghi
Chemical Science (2010-Present) 2012 - vol. 3(Issue 6) pp:NaN1803-1803
Publication Date(Web):2012/03/30
DOI:10.1039/C2SC20270K
The first total synthesis of (+)-gliocladin B is described. Our concise and enantioselective synthesis takes advantage of a new regioselective Friedel–Crafts-based strategy to provide an efficient multigram-scale access to the C3-(3′-indolyl)hexahydropyrroloindole substructure, a molecular foundation present in a significant subset of epipolythiodiketopiperazine natural alkaloids. Our first-generation solution to (+)-gliocladin B involved the stereoselective formation of (+)-12-deoxybionectin A, a plausible biosynthetic precursor. Our synthesis clarified the C15 stereochemistry of (+)-gliocladin B and allowed its full structure confirmation. Further studies of a versatile dihydroxylated diketopiperazine provided a concise and efficient synthesis of (+)-gliocladin B as well as access to (+)-gliocladin C.
Co-reporter:Alison E. Ondrus and Mohammad Movassaghi
Chemical Communications 2009(Issue 28) pp:NaN4165-4165
Publication Date(Web):2009/05/19
DOI:10.1039/B903995N
The myrmicarins are a family of air- and temperature-sensitive alkaloids that possess unique structural features. Our concise enantioselective synthesis of the tricyclic myrmicarins enabled evaluation of a potentially biomimetic assembly of the complex members via direct dimerization of simpler structures. These studies revealed that myrmicarin 215B undergoes efficient and highly diastereoselective Brønsted acid-induced dimerization to generate a new heptacyclic structure, isomyrmicarin 430A. Mechanistic analysis demonstrated that heterodimerization between myrmicarin 215B and a conformationally restricted azafulvenium ion precursor afforded a functionalized isomyrmicarin 430A structure in a manner that was consistent with a highly efficient, non-concerted ionic process. Recent advancement in heterodimerization between tricyclic derivatives has enabled the preparation of strategically functionalized hexacyclic structures. The design and synthesis of structurally versatile dimeric compounds has greatly facilitated manipulation of these structures en route to more complex myrmicarin derivatives.
Co-reporter:Jonathan William Medley and Mohammad Movassaghi
Chemical Communications 2013 - vol. 49(Issue 92) pp:NaN10777-10777
Publication Date(Web):2013/09/27
DOI:10.1039/C3CC44461A
The 1917 total synthesis of tropinone by Sir Robert Robinson represents a landmark achievement in organic synthesis. Decades ahead of its time in terms of its retrosynthetic logic and biomimetic approach, the elegant combination of these two elements in this synthesis continues to serve as an inspiration for the development of new and efficient strategies for complex molecule synthesis.
.jpg)
![Dinaphtho[2,1-d:1',2'-f][1,3,2]dioxaphosphepin,4-hydroxy-2,6-bis[2,4,6-tris(1-methylethyl)phenyl]-, 4-oxide, (11bS)-](/data/chemimg/103700/874948-63-7.png)
![Dinaphtho[2,1-d:1',2'-f][1,3,2]dioxaphosphepin,4-hydroxy-2,6-bis[2,4,6-tris(1-methylethyl)phenyl]-, 4-oxide, (11bS)-](/data/chemimg/103700/874948-63-7_b.png)
![Rhodium, bis[m-[a,a,a',a'-tetramethyl-1,3-benzenedipropanoato(2-)-kO1,kO'3:kO3,kO'1]]di-, (Rh-Rh)](http://img.cochemist.com/ccimg/819100/819050-89-0.png)
![Rhodium, bis[m-[a,a,a',a'-tetramethyl-1,3-benzenedipropanoato(2-)-kO1,kO'3:kO3,kO'1]]di-, (Rh-Rh)](http://img.cochemist.com/ccimg/819100/819050-89-0_b.png)
![2l5-1,3,2-Diazaphosphorin-2-amine,2-[(1,1-dimethylethyl)imino]-N,N-diethylhexahydro-1,3-dimethyl-](http://img.cochemist.com/ccimg/98100/98015-45-3.png)
![2l5-1,3,2-Diazaphosphorin-2-amine,2-[(1,1-dimethylethyl)imino]-N,N-diethylhexahydro-1,3-dimethyl-](http://img.cochemist.com/ccimg/98100/98015-45-3_b.png)
![Carbamic acid, [2-(4-bromo-1H-indol-3-yl)ethyl]-, 1,1-dimethylethylester](http://img.cochemist.com/ccimg/217500/217493-25-9.png)
![Carbamic acid, [2-(4-bromo-1H-indol-3-yl)ethyl]-, 1,1-dimethylethylester](http://img.cochemist.com/ccimg/217500/217493-25-9_b.png)


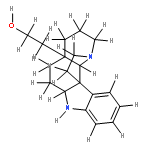
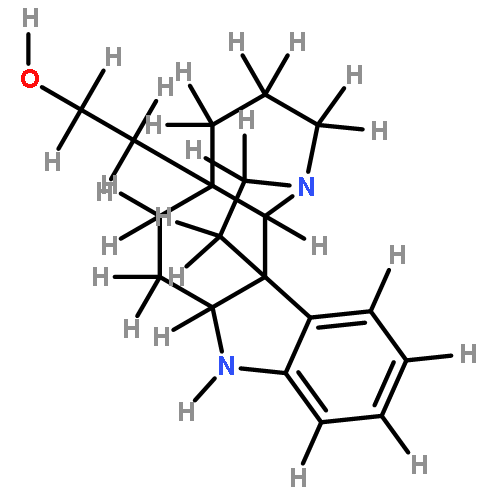
![CARBAMIC ACID, [2-(1H-INDOL-3-YL)ETHYL]-, PHENYLMETHYL ESTER](http://img.cochemist.com/ccimg/38800/38750-13-9.png)
![CARBAMIC ACID, [2-(1H-INDOL-3-YL)ETHYL]-, PHENYLMETHYL ESTER](http://img.cochemist.com/ccimg/38800/38750-13-9_b.png)
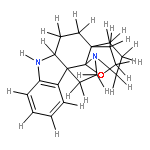
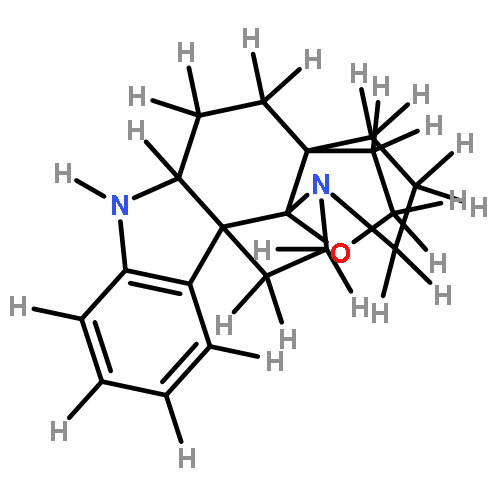
![Acetamide, N-[2-[1-[(4-methylphenyl)sulfonyl]-1H-indol-3-yl]ethyl]-](http://img.cochemist.com/ccimg/919800/919787-20-5.png)
![Acetamide, N-[2-[1-[(4-methylphenyl)sulfonyl]-1H-indol-3-yl]ethyl]-](http://img.cochemist.com/ccimg/919800/919787-20-5_b.png)
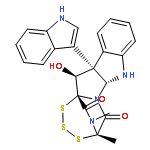
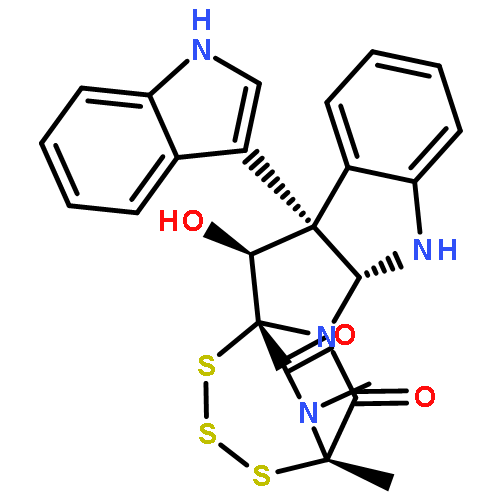
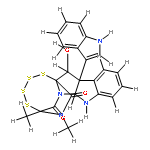
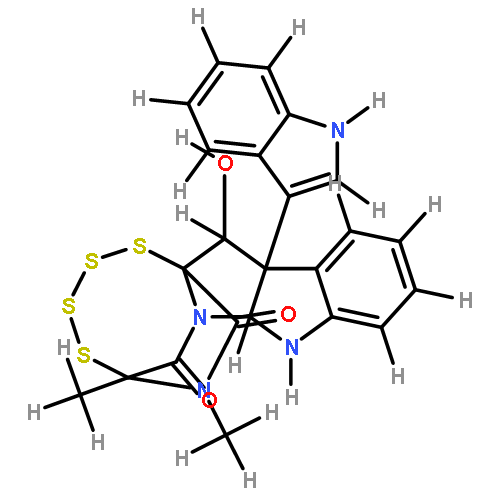
![BENZENESULFONAMIDE, N-[2-(1H-INDOL-3-YL)ETHYL]-N,2,4,6-TETRAMETHYL-](http://img.cochemist.com/ccimg/668500/668419-46-3.png)
![BENZENESULFONAMIDE, N-[2-(1H-INDOL-3-YL)ETHYL]-N,2,4,6-TETRAMETHYL-](http://img.cochemist.com/ccimg/668500/668419-46-3_b.png)



![L-Serine, O-[(1,1-dimethylethyl)diphenylsilyl]-, methyl ester](/data/chemimg/1109900/140408-49-7.png)
![L-Serine, O-[(1,1-dimethylethyl)diphenylsilyl]-, methyl ester](/data/chemimg/1109900/140408-49-7_b.png)
![2-Butenal, 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-, (2Z)-](http://img.cochemist.com/ccimg/127400/127382-61-0.png)
![2-Butenal, 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-, (2Z)-](http://img.cochemist.com/ccimg/127400/127382-61-0_b.png)
![1H-Indole,1-[tris(1-methylethyl)silyl]-](http://img.cochemist.com/ccimg/123200/123191-00-4.png)
![1H-Indole,1-[tris(1-methylethyl)silyl]-](http://img.cochemist.com/ccimg/123200/123191-00-4_b.png)
![2-Butenal, 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-, (2E)-](http://img.cochemist.com/ccimg/119200/119125-30-3.png)
![2-Butenal, 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-, (2E)-](http://img.cochemist.com/ccimg/119200/119125-30-3_b.png)
![4-[TERT-BUTYL(DIMETHYL)SILYL]OXYBUT-2-YNAL](http://img.cochemist.com/ccimg/75900/75859-97-1.png)
![4-[TERT-BUTYL(DIMETHYL)SILYL]OXYBUT-2-YNAL](http://img.cochemist.com/ccimg/75900/75859-97-1_b.png)
![Carbamic acid, [2-(1H-indol-3-yl)ethyl]-, methyl ester](http://img.cochemist.com/ccimg/58700/58635-45-3.png)
![Carbamic acid, [2-(1H-indol-3-yl)ethyl]-, methyl ester](http://img.cochemist.com/ccimg/58700/58635-45-3_b.png)
![1-[2-(1-METHYLINDOL-3-YL)ETHYL]PIPERIDIN-2-ONE](http://img.cochemist.com/ccimg/56200/56186-10-8.png)
![1-[2-(1-METHYLINDOL-3-YL)ETHYL]PIPERIDIN-2-ONE](http://img.cochemist.com/ccimg/56200/56186-10-8_b.png)
![ACETAMIDE, N-[2-[1-(PHENYLMETHYL)-1H-INDOL-3-YL]ETHYL]-](http://img.cochemist.com/ccimg/41700/41645-07-2.png)
![ACETAMIDE, N-[2-[1-(PHENYLMETHYL)-1H-INDOL-3-YL]ETHYL]-](http://img.cochemist.com/ccimg/41700/41645-07-2_b.png)
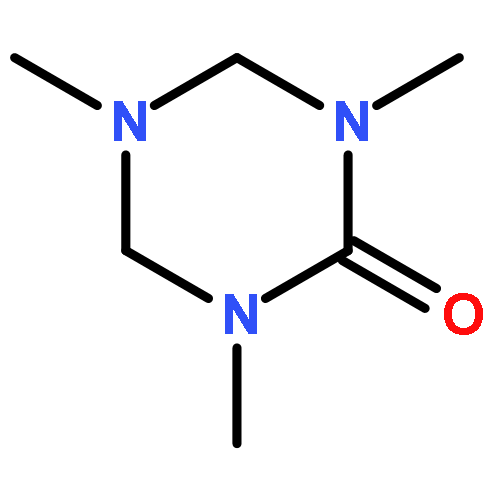
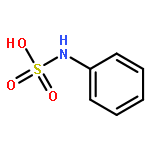
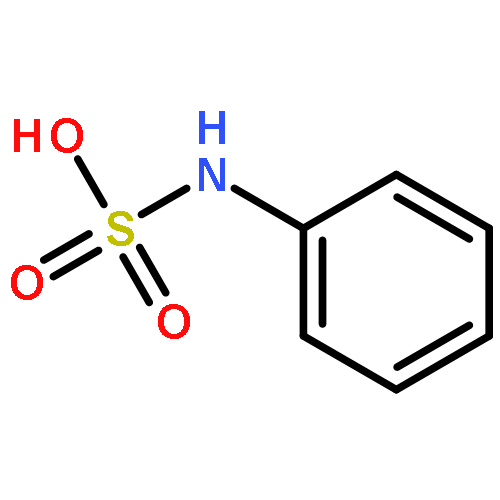
![Propanamide, N-[2-(1H-indol-3-yl)ethyl]-2,2-dimethyl-](http://img.cochemist.com/ccimg/15800/15776-48-4.png)
![Propanamide, N-[2-(1H-indol-3-yl)ethyl]-2,2-dimethyl-](http://img.cochemist.com/ccimg/15800/15776-48-4_b.png)
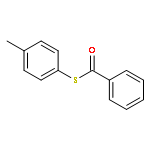
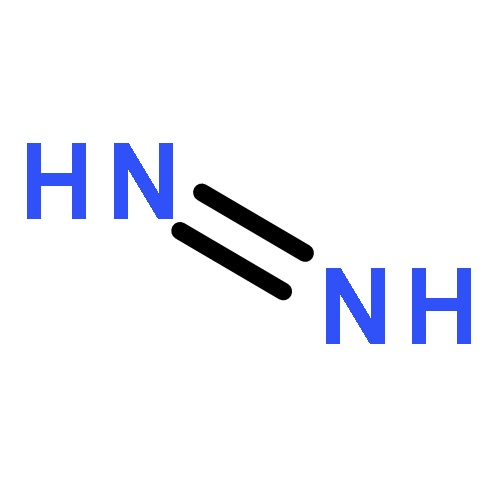
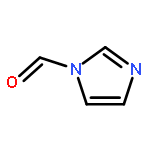
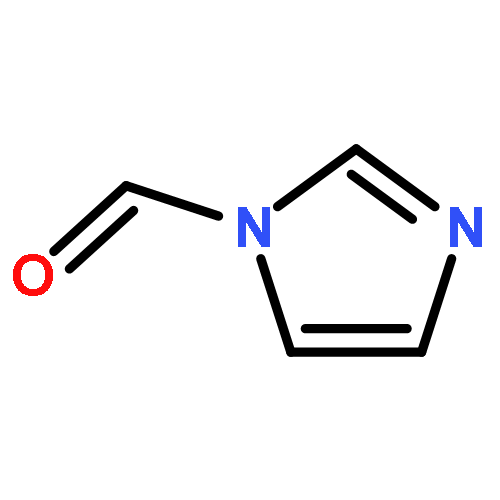
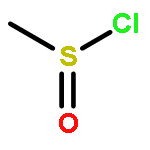
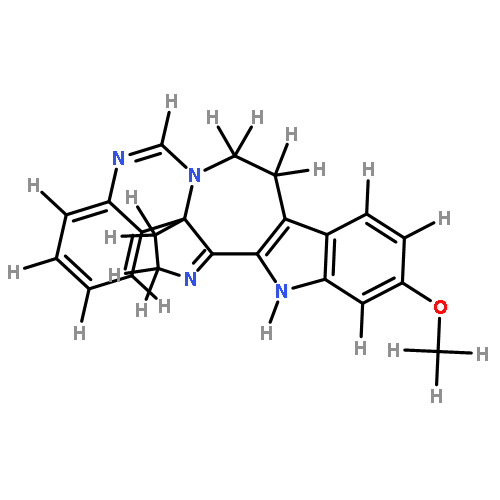
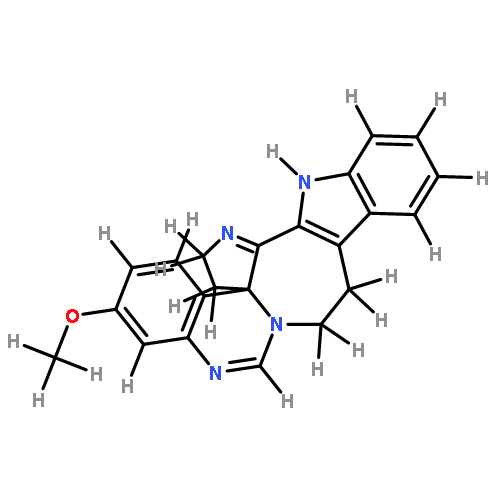
![Formamide, N-[2-[(12bS)-6,7,12,13-tetrahydro-2-methoxy-12bH-azepino[3,2-b:4,5-b']diindol-12b-yl]ethyl]-](/data/chemimg/168500/1227157-89-2.png)
![Formamide, N-[2-[(12bS)-6,7,12,13-tetrahydro-2-methoxy-12bH-azepino[3,2-b:4,5-b']diindol-12b-yl]ethyl]-](/data/chemimg/168500/1227157-89-2_b.png)
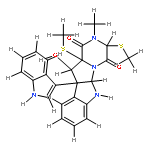
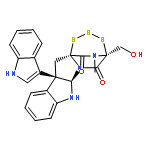
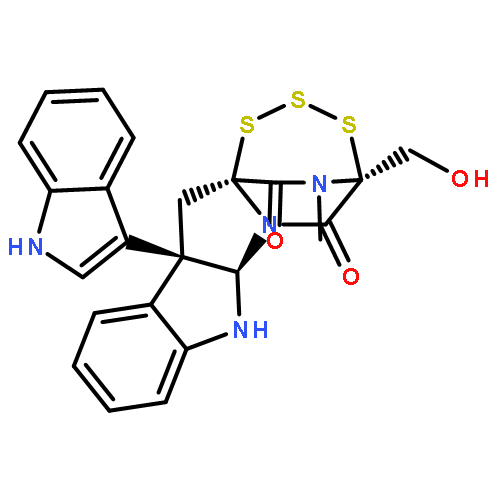
![[11b,11'b(12H,12'H)-Bi-4,12a-(iminomethano)-12aH-[1,2,3,5]trithiazepino[5',4':1,5]pyrrolo[2,3-b]indole]-5,5',13,13'(4H,4'H)-tetrone,6a,6'a,7,7'-tetrahydro-4,4'-bis(hydroxymethyl)-14,14'-dimethyl-,(4S,4'S,6aR,6'aR,11bR,11'bR,12aS,12'aS)- (9CI)](http://img.cochemist.com/ccimg/118200/118111-08-3.png)
![[11b,11'b(12H,12'H)-Bi-4,12a-(iminomethano)-12aH-[1,2,3,5]trithiazepino[5',4':1,5]pyrrolo[2,3-b]indole]-5,5',13,13'(4H,4'H)-tetrone,6a,6'a,7,7'-tetrahydro-4,4'-bis(hydroxymethyl)-14,14'-dimethyl-,(4S,4'S,6aR,6'aR,11bR,11'bR,12aS,12'aS)- (9CI)](http://img.cochemist.com/ccimg/118200/118111-08-3_b.png)
![1,1',1''-[(chlorodisulfanyl)methanetriyl]tribenzene](http://img.cochemist.com/ccimg/35600/35572-83-9.png)
![1,1',1''-[(chlorodisulfanyl)methanetriyl]tribenzene](http://img.cochemist.com/ccimg/35600/35572-83-9_b.png)
![1H-Isoindole-1,3(2H)-dione,2-[2-(1H-indol-3-yl)ethyl]-](http://img.cochemist.com/ccimg/15800/15741-71-6.png)
![1H-Isoindole-1,3(2H)-dione,2-[2-(1H-indol-3-yl)ethyl]-](http://img.cochemist.com/ccimg/15800/15741-71-6_b.png)
![(5aS,5bS,8aS,9aR)-1-bromo-8a-hydroxy-5,5a,5b,6,8,8a,9,9a-octahydroimidazo[4',5':4,5]cyclopenta[1,2-e]pyrrolo[1,2-a]pyrazine-4,7-dione](http://img.cochemist.com/ccimg/201400/201338-45-6.png)
![(5aS,5bS,8aS,9aR)-1-bromo-8a-hydroxy-5,5a,5b,6,8,8a,9,9a-octahydroimidazo[4',5':4,5]cyclopenta[1,2-e]pyrrolo[1,2-a]pyrazine-4,7-dione](http://img.cochemist.com/ccimg/201400/201338-45-6_b.png)
![Imidazo[4',5':4,5]cyclopenta[1,2-e]pyrrolo[1,2-a]pyrazine-4,7-dione,1-bromo-5,5a,5b,6,8,8a,9,9a-octahydro-5b,8a-dihydroxy-8-methyl-,(5aR,5bR,8aS,9aR)-](http://img.cochemist.com/ccimg/201400/201338-44-5.png)
![Imidazo[4',5':4,5]cyclopenta[1,2-e]pyrrolo[1,2-a]pyrazine-4,7-dione,1-bromo-5,5a,5b,6,8,8a,9,9a-octahydro-5b,8a-dihydroxy-8-methyl-,(5aR,5bR,8aS,9aR)-](http://img.cochemist.com/ccimg/201400/201338-44-5_b.png)
![Imidazo[4',5':4,5]cyclopenta[1,2-e]pyrrolo[1,2-a]pyrazine-4,7-dione,1-bromo-5,5a,5b,6,8,8a,9,9a-octahydro-8a-hydroxy-8-methyl-, (5aS,5bS,8aS,9aR)-](http://img.cochemist.com/ccimg/152500/152406-28-5.png)
![Imidazo[4',5':4,5]cyclopenta[1,2-e]pyrrolo[1,2-a]pyrazine-4,7-dione,1-bromo-5,5a,5b,6,8,8a,9,9a-octahydro-8a-hydroxy-8-methyl-, (5aS,5bS,8aS,9aR)-](http://img.cochemist.com/ccimg/152500/152406-28-5_b.png)
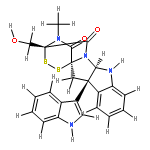
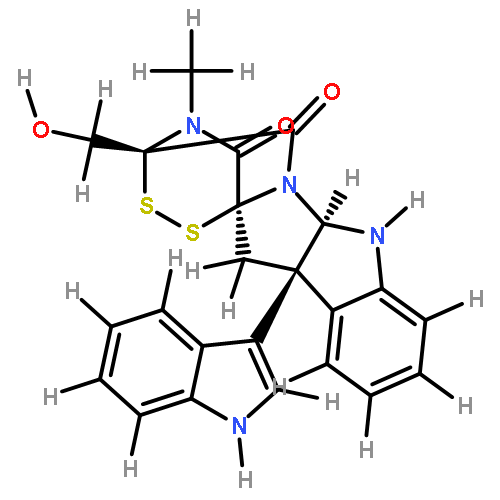
![[10b,10'b(11H,11'H)-Bi-3,11a-epidithio-11aH-pyrazino[1',2':1,5]pyrrolo[2,3-b]indole]-1,1',4,4'-tetrone,2,2',3,3',5a,5'a,6,6'-octahydro-3,3'-bis(hydroxymethyl)-2,2'-dimethyl-,(3S,3'S,5aR,5'aR,10bR,10'bR,11aS,11'aS)-](http://img.cochemist.com/ccimg/28100/28097-03-2.png)
![[10b,10'b(11H,11'H)-Bi-3,11a-epidithio-11aH-pyrazino[1',2':1,5]pyrrolo[2,3-b]indole]-1,1',4,4'-tetrone,2,2',3,3',5a,5'a,6,6'-octahydro-3,3'-bis(hydroxymethyl)-2,2'-dimethyl-,(3S,3'S,5aR,5'aR,10bR,10'bR,11aS,11'aS)-](http://img.cochemist.com/ccimg/28100/28097-03-2_b.png)