Co-reporter:Dr. Priya R. Banerjee;Anthony N. Milin;Dr. Mahdi Muhammad Moosa;Paulo L. Onuchic; Ashok A. Deniz
Angewandte Chemie 2017 Volume 129(Issue 38) pp:
Publication Date(Web):2017/09/11
DOI:10.1002/ange.201783861
PhasenumwandlungIn der Zuschrift auf S. 11512 zeigen P. R. Banerjee, A. A. Deniz et al., dass die Bildung und Auflösung von Ribonucleoprotein-basierten organellartigen Tröpfchen mittels RNA kontrolliert werden kann.
Co-reporter:Dr. Priya R. Banerjee;Anthony N. Milin;Dr. Mahdi Muhammad Moosa;Paulo L. Onuchic; Ashok A. Deniz
Angewandte Chemie 2017 Volume 129(Issue 38) pp:11512-11517
Publication Date(Web):2017/09/11
DOI:10.1002/ange.201703191
AbstractIntracellular ribonucleoprotein (RNP) granules are membrane-less droplet organelles that are thought to regulate posttranscriptional gene expression. While liquid–liquid phase separation may drive RNP granule assembly, the mechanisms underlying their supramolecular dynamics and internal organization remain poorly understood. Herein, we demonstrate that RNA, a primary component of RNP granules, can modulate the phase behavior of RNPs by controlling both droplet assembly and dissolution in vitro. Monotonically increasing the RNA concentration initially leads to droplet assembly by complex coacervation and subsequently triggers an RNP charge inversion, which promotes disassembly. This RNA-mediated reentrant phase transition can drive the formation of dynamic droplet substructures (vacuoles) with tunable lifetimes. We propose that active cellular processes that can create an influx of RNA into RNP granules, such as transcription, can spatiotemporally control the organization and dynamics of such liquid-like organelles.
Co-reporter:Mahdi Muhammad Moosa;Dr. Allan Chris M. Ferreon; Dr. Ashok A. Deniz
ChemPhysChem 2015 Volume 16( Issue 1) pp:90-94
Publication Date(Web):
DOI:10.1002/cphc.201402661
Abstract
Intrinsically disordered proteins (IDPs) are involved in diverse cellular functions. Many IDPs can interact with multiple binding partners, resulting in their folding into alternative ligand-specific functional structures. For such multi-structural IDPs, a key question is whether these multiple structures are fully encoded in the protein sequence, as is the case in many globular proteins. To answer this question, here we employed a combination of single-molecule and ensemble techniques to compare ligand-induced and osmolyte-forced folding of α-synuclein. Our results reveal context-dependent modulation of the protein′s folding landscape, suggesting that the codes for the protein′s native folds are partially encoded in its primary sequence, and are completed only upon interaction with binding partners. Our findings suggest a critical role for cellular interactions in expanding the repertoire of folds and functions available to disordered proteins.
Co-reporter:Priya R. Banerjee and Ashok A. Deniz
Chemical Society Reviews 2014 vol. 43(Issue 4) pp:1172-1188
Publication Date(Web):16 Dec 2013
DOI:10.1039/C3CS60311C
Single-molecule (SM) fluorescence methods have been increasingly instrumental in our current understanding of a number of key aspects of protein folding and aggregation landscapes over the past decade. With the advantage of a model free approach and the power of probing multiple subpopulations and stochastic dynamics directly in a heterogeneous structural ensemble, SM methods have emerged as a principle technique for studying complex systems such as intrinsically disordered proteins (IDPs), globular proteins in the unfolded basin and during folding, and early steps of protein aggregation in amyloidogenesis. This review highlights the application of these methods in investigating the free energy landscapes, folding properties and dynamics of individual protein molecules and their complexes, with an emphasis on inherently flexible systems such as IDPs.
Co-reporter:Rajaraman Krishnan;Jessica L. Goodman;Samrat Mukhopadhyay;Chris D. Pacheco;Edward A. Lemke;Susan Lindquist
PNAS 2012 109 (28 ) pp:
Publication Date(Web):2012-07-10
DOI:10.1073/pnas.1209527109
Some amyloid-forming polypeptides are associated with devastating human diseases and others provide important biological functions.
For both, oligomeric intermediates appear during amyloid assembly. Currently we have few tools for characterizing these conformationally
labile intermediates and discerning what governs their benign versus toxic states. Here, we examine intermediates in the assembly
of a normal, functional amyloid, the prion-determining region of yeast Sup35 (NM). During assembly, NM formed a variety of
oligomers with different sizes and conformation-specific antibody reactivities. Earlier oligomers were less compact and reacted
with the conformational antibody A11. More mature oligomers were more compact and reacted with conformational antibody OC.
We found we could arrest NM in either of these two distinct oligomeric states with small molecules or crosslinking. The A11-reactive
oligomers were more hydrophobic (as measured by Nile Red binding) and were highly toxic to neuronal cells, while OC-reactive
oligomers were less hydrophobic and were not toxic. The A11 and OC antibodies were originally raised against oligomers of
Aβ, an amyloidogenic peptide implicated in Alzheimer’s disease (AD) that is completely unrelated to NM in sequence. Thus,
this natural yeast prion samples two conformational states similar to those sampled by Aβ, and when assembly stalls at one
of these two states, but not the other, it becomes extremely toxic. Our results have implications for selective pressures
operating on the evolution of amyloid folds across a billion years of evolution. Understanding the features that govern such
conformational transitions will shed light on human disease and evolution alike.
Co-reporter:Allan Chris M. Ferreon;Yann Gambin;Mahdi Muhammad Moosa
PNAS 2012 Volume 109 (Issue 44 ) pp:
Publication Date(Web):2012-10-30
DOI:10.1073/pnas.1201802109
Protein structure and function depend on a close interplay between intrinsic folding energy landscapes and the chemistry of
the protein environment. Osmolytes are small-molecule compounds that can act as chemical chaperones by altering the environment
in a cellular context. Despite their importance, detailed studies on the role of these chemical chaperones in modulating structure
and dimensions of intrinsically disordered proteins have been limited. Here, we used single-molecule Förster resonance energy
transfer to test the counteraction hypothesis of counterbalancing effects between the protecting osmolyte trimethylamine-N-oxide
(TMAO) and denaturing osmolyte urea for the case of α-synuclein, a Parkinson’s disease-linked protein whose monomer exhibits
significant disorder. The single-molecule experiments, which avoid complications from protein aggregation, do not exhibit
clear solvent-induced cooperative protein transitions for these osmolytes, unlike results from previous studies on globular
proteins. Our data demonstrate the ability of TMAO and urea to shift α-synuclein structures towards either more compact or
expanded average dimensions. Strikingly, the experiments directly reveal that a 2∶1 [urea]∶[TMAO] ratio has a net neutral
effect on the protein’s dimensions, a result that holds regardless of the absolute osmolyte concentrations. Our findings shed
light on a surprisingly simple aspect of the interplay between urea and TMAO on α-synuclein in the context of intrinsically
disordered proteins, with potential implications for the biological roles of such chemical chaperones. The results also highlight
the strengths of single-molecule experiments in directly probing the chemical physics of protein structure and disorder in
more chemically complex environments.
Co-reporter:Yann Gambin and Ashok A. Deniz
Molecular BioSystems 2010 vol. 6(Issue 9) pp:1540-1547
Publication Date(Web):02 Jul 2010
DOI:10.1039/C003024D
Proper protein function in cells, tissues and organisms depends critically on correct protein folding or interaction with partners. Over the last decade, single-molecule FRET (smFRET) has emerged as a powerful tool to probe complex distributions, dynamics, pathways and landscapes in protein folding and binding reactions, leveraging its ability to avoid averaging over an ensemble of molecules. While smFRET was practiced in a two-color form until recently, the last few years have seen the development of enhanced multicolor smFRET methods that provide additional structural information permitting us to probe more complex mechanisms. In this review, we provide a brief introduction to the smFRET technique, then follow with advanced multicolor measurements and end with ongoing methodology developments in microfluidics and protein labeling that are beginning to make these techniques more broadly applicable to answering a number of key questions about folding and binding.
Co-reporter:AllanChrisM. Ferreon Dr.;CrystalR. Moran;JosephineC. Ferreon Dr. ;AshokA. Deniz Dr.
Angewandte Chemie 2010 Volume 122( Issue 20) pp:3547-3550
Publication Date(Web):
DOI:10.1002/ange.201000378
Co-reporter:AllanChrisM. Ferreon Dr.;CrystalR. Moran;JosephineC. Ferreon Dr. ;AshokA. Deniz Dr.
Angewandte Chemie International Edition 2010 Volume 49( Issue 20) pp:3469-3472
Publication Date(Web):
DOI:10.1002/anie.201000378
Co-reporter:Edward A. Lemke ; Yann Gambin ; Virginia Vandelinder ; Eric M. Brustad ; Hsiao-Wei Liu ; Peter G. Schultz ; Alex Groisman
Journal of the American Chemical Society 2009 Volume 131(Issue 38) pp:13610-13612
Publication Date(Web):September 4, 2009
DOI:10.1021/ja9027023
A microfluidic device made of polydimethylsiloxane (PDMS) addresses key limitations in single-molecule fluorescence experiments by providing high dye photostability and low sample sticking. Photobleaching is dramatically reduced by deoxygenation via gas diffusion through porous channel walls. Rapid buffer exchange in a laminar sheath flow followed by optical interrogation minimizes surface−sample contacts and allows the in situ addition and combination of other reagents.
Co-reporter:Virginia Vandelinder, Allan Chris M. Ferreon, Yann Gambin, Ashok A. Deniz and Alex Groisman
Analytical Chemistry 2009 Volume 81(Issue 16) pp:6929
Publication Date(Web):June 25, 2009
DOI:10.1021/ac901008c
We present a microfluidic device for rapid and efficient determination of protein conformations in a range of medium conditions and temperatures. The device generates orthogonal gradients of concentration and temperature in an interrogation area that fits into the field of view of an objective lens with a numerical aperture of 0.45. A single Förster resonance energy transfer (FRET) image of the interrogation area containing a dual-labeled protein provides a 100 × 100 point map of the FRET efficiency that corresponds to a diagram of protein conformations in the coordinates of temperature and medium conditions. The device is used to explore the conformations of α-synuclein, an intrinsically disordered protein linked to Parkinson’s and Alzheimer’s diseases, in the presence of a binding partner, the lipid-mimetic sodium dodecyl sulfate (SDS). The experiment provides a diagram of conformations of α-synuclein with 10 000 individual data points in a range of 21−47 °C and 0−2.5 mM SDS. The diagram is consistent with previous reports but also reveals new conformational transitions that would be difficult to detect with conventional techniques. The microfluidic device can potentially be used to study other biomolecular and soft-matter systems.
Co-reporter:Allan Chris M. Ferreon;Yann Gambin;Edward A. Lemke
PNAS 2009 Volume 106 (Issue 14 ) pp:5645-5650
Publication Date(Web):2009-04-07
DOI:10.1073/pnas.0809232106
We studied the coupled binding and folding of α-synuclein, an intrinsically disordered protein linked with Parkinson's disease.
Using single-molecule fluorescence resonance energy transfer and correlation methods, we directly probed protein membrane
association, structural distributions, and dynamics. Results revealed an intricate energy landscape on which binding of α-synuclein
to amphiphilic small molecules or membrane-like partners modulates conformational transitions between a natively unfolded
state and multiple α-helical structures. α-Synuclein conformation is not continuously tunable, but instead partitions into
2 main classes of folding landscape structural minima. The switch between a broken and an extended helical structure can be
triggered by changing the concentration of binding partners or by varying the curvature of the binding surfaces presented
by micelles or bilayers composed of the lipid-mimetic SDS. Single-molecule experiments with lipid vesicles of various composition
showed that a low fraction of negatively charged lipids, similar to that found in biological membranes, was sufficient to
drive α-synuclein binding and folding, resulting here in the induction of an extended helical structure. Overall, our results
imply that the 2 folded structures are preencoded by the α-synuclein amino acid sequence, and are tunable by small-molecule
supramolecular states and differing membrane properties, suggesting novel control elements for biological and amyloid regulation
of α-synuclein.
Co-reporter:Yann Gambin;Alexander Schug;Thomas J. Magliery;Edward A. Lemke;Jason J. Lavinder;Allan Chris M. Ferreon;José N. Onuchic
PNAS 2009 Volume 106 (Issue 25 ) pp:10153-10158
Publication Date(Web):2009-06-23
DOI:10.1073/pnas.0904461106
Biological activity in proteins requires them to share the energy landscape for folding and global conformational motions,
2 key determinants of function. Although most structural studies to date have focused on fluctuations around a single structural
basin, we directly observe the coexistence of 2 symmetrically opposed conformations for a mutant of the Rop-homodimer (Repressor
of Primer) in single-molecule fluorescence resonance energy transfer (smFRET) measurements. We find that mild denaturing conditions
can affect the sensitive balance between the conformations, generating an equilibrium ensemble consisting of 2 equally occupied
structural basins. Despite the need for large-scale conformational rearrangement, both native structures are dynamically and
reversibly adopted for the same paired molecules without separation of the constituent monomers. Such an ability of some proteins
or protein complexes to switch between conformations by thermal fluctuations and/or minor environmental changes could be central
to their ability to control biological function.
Co-reporter:Samrat Mukhopadhyay;Rajaraman Krishnan;Edward A. Lemke;Susan Lindquist
PNAS 2007 Volume 104 (Issue 8 ) pp:2649-2654
Publication Date(Web):2007-02-20
DOI:10.1073/pnas.0611503104
The yeast prion protein Sup35 is a translation termination factor, whose activity is modulated by sequestration into a self-perpetuating
amyloid. The prion-determining domain, NM, consists of two distinct regions: an amyloidogenic N terminus domain (N) and a
charged solubilizing middle region (M). To gain insight into prion conversion, we used single-molecule fluorescence resonance
energy transfer (SM-FRET) and fluorescence correlation spectroscopy to investigate the structure and dynamics of monomeric
NM. Low protein concentrations in these experiments prevented the formation of obligate on-pathway oligomers, allowing us
to study early folding intermediates in isolation from higher-order species. SM-FRET experiments on a dual-labeled amyloid
core variant (N21C/S121C, retaining wild-type prion behavior) indicated that the N region of NM adopts a collapsed form similar
to “burst-phase” intermediates formed during the folding of many globular proteins, even though it lacks a typical hydrophobic
core. The mean distance between residues 21 and 121 was ≈43 Å. This increased with denaturant in a noncooperative fashion
to ≈63 Å, suggesting a multitude of interconverting species rather than a small number of discrete monomeric conformers. Fluorescence
correlation spectroscopy analysis of singly labeled NM revealed fast conformational fluctuations on the 20- to 300-ns time
scale. Quenching from proximal and distal tyrosines resulted in distinct fast and slower fluctuations. Our results indicate
that native monomeric NM is composed of an ensemble of structures, having a collapsed and rapidly fluctuating N region juxtaposed
with a more extended M region. The stability of such ensembles is likely to play a key role in prion conversion.
Co-reporter:Svitlana Y. Berezhna;Lubica Supekova;Frantisek Supek;Peter G. Schultz
PNAS 2006 Volume 103 (Issue 20 ) pp:7682-7687
Publication Date(Web):2006-05-16
DOI:10.1073/pnas.0600148103
Recent observations of RNA interference (RNAi) in the nuclei of human cells raise key questions about the extent to which
nuclear and cytoplasmic RNAi pathways are shared. By directly visualizing the localization of small interfering RNA (siRNA)
in live human cells, we show here that siRNA either selectively localizes in the cytoplasm or translocates into the nucleus,
depending on where the silencing target RNA resides. Two siRNAs that target the small nuclear 7SK and U6 RNAs localize into
the nucleus as duplexes. In contrast, an siRNA targeting the cytoplasmic hepatitis C virus replicon RNA dissociates, and only
antisense strand distributes in the cytoplasm of the cells harboring the target RNA, whereas sense strand gets degraded. At
the same time, both strands of the latter siRNA are distributed throughout the cytoplasm and nucleus in cells lacking the
silencing target RNA. These results suggest the existence of a mechanism by which the RNAi machinery orchestrates a target-determined
localization of the siRNA and the corresponding RNAi activity, and also provide evidence for formation of nuclear-programmed
active RNA induced silencing complexes directly in the nucleus.
Co-reporter:Jean-Pierre Clamme Dr.
ChemPhysChem 2005 Volume 6(Issue 1) pp:
Publication Date(Web):13 DEC 2004
DOI:10.1002/cphc.200400261
A molecular ruler: A three-color single-molecule fluorescence resonance energy transfer (FRET) method is presented (see Figure inset), which can be used to simultaneously measure multiple molecular distance changes during molecular conformational changes and binding. The ability to directly study conformational subpopulations in a mixture of molecules with different interdye distances is highlighted by the well-separated peaks in the two- dimensional histogram in the Figure.
Co-reporter:Taehyung Lee, Crystal R. Moran-Gutierrez, Ashok A. Deniz
Seminars in Cell & Developmental Biology (January 2015) Volume 37() pp:26-34
Publication Date(Web):1 January 2015
DOI:10.1016/j.semcdb.2014.09.027
A substantial fraction of the human proteome encodes disordered proteins. Protein disorder is associated with a variety of cellular functions and misfunction, and is therefore of clear import to biological systems. However, disorder lends itself to conformational flexibility and heterogeneity, rendering proteins which feature prominent disorder difficult to study using conventional structural biology methods. Here we discuss a few examples of how single-molecule methods are providing new insight into the biophysics and complexity of these proteins by avoiding ensemble averaging, thereby providing direct information about the complex distributions and dynamics of this important class of proteins. Examples of note include characterization of isolated IDPs in solution as collapsed and dynamic species, detailed insight into complex IDP folding landscapes, and new information about how tunable regulation of structure-mediated binding cooperativity and consequent function can be achieved through protein disorder. With these exciting advances in view, we conclude with a discussion of a few complementary and emerging single-molecule efforts of particular promise, including complementary and enhanced methodologies for studying disorder in proteins, and experiments to investigate the potential role for IDP-induced phase separation as a critical functional element in biological systems.
Co-reporter:Priya R. Banerjee and Ashok A. Deniz
Chemical Society Reviews 2014 - vol. 43(Issue 4) pp:NaN1188-1188
Publication Date(Web):2013/12/16
DOI:10.1039/C3CS60311C
Single-molecule (SM) fluorescence methods have been increasingly instrumental in our current understanding of a number of key aspects of protein folding and aggregation landscapes over the past decade. With the advantage of a model free approach and the power of probing multiple subpopulations and stochastic dynamics directly in a heterogeneous structural ensemble, SM methods have emerged as a principle technique for studying complex systems such as intrinsically disordered proteins (IDPs), globular proteins in the unfolded basin and during folding, and early steps of protein aggregation in amyloidogenesis. This review highlights the application of these methods in investigating the free energy landscapes, folding properties and dynamics of individual protein molecules and their complexes, with an emphasis on inherently flexible systems such as IDPs.
.jpg)









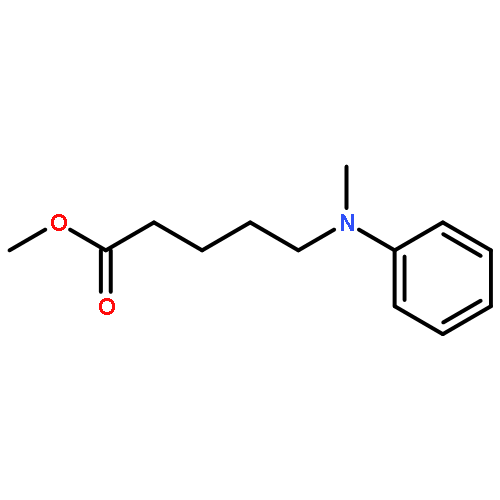
![Pentanoic acid, 5-[(4-formylphenyl)methylamino]-, methyl ester](http://img.cochemist.com/ccimg/117900/117846-69-2.png)
![Pentanoic acid, 5-[(4-formylphenyl)methylamino]-, methyl ester](http://img.cochemist.com/ccimg/117900/117846-69-2_b.png)




![Benzonitrile, 4-[2-[4-(dimethylamino)phenyl]ethenyl]-, (E)-](http://img.cochemist.com/ccimg/2900/2844-17-9.png)
![Benzonitrile, 4-[2-[4-(dimethylamino)phenyl]ethenyl]-, (E)-](http://img.cochemist.com/ccimg/2900/2844-17-9_b.png)


![3',6'-Dihydroxy-3H-spiro[isobenzofuran-1,9'-xanthen]-3-one](http://img.cochemist.com/ccimg/2400/2321-07-5.png)
![3',6'-Dihydroxy-3H-spiro[isobenzofuran-1,9'-xanthen]-3-one](http://img.cochemist.com/ccimg/2400/2321-07-5_b.png)

