Co-reporter:Daniel Mieritz, Xiang Li, Alex Volosin, Minghui Liu, Hao Yan, Nils G. Walter, and Dong-Kyun Seo
Langmuir June 27, 2017 Volume 33(Issue 25) pp:6410-6410
Publication Date(Web):June 2, 2017
DOI:10.1021/acs.langmuir.7b00761
Housing bio-nano guest devices based on DNA nanostructures within porous, conducting, inorganic host materials promise valuable applications in solar energy conversion, chemical catalysis, and analyte sensing. Herein, we report a single-template synthetic development of hierarchically porous, transparent conductive metal oxide coatings whose pores are freely accessible by large biomacromolecules. Their hierarchal pore structure is bimodal with a larger number of closely packed open macropores (∼200 nm) at the higher rank and with the remaining space being filled with a gel network of antimony-doped tin oxide (ATO) nanoparticles that is highly porous with a broad size range of textual pores mainly from 20–100 nm at the lower rank. The employed carbon black template not only creates the large open macropores but also retains the highly structured gel network as holey pore walls. Single molecule fluorescence microscopic studies with fluorophore-labeled DNA nanotweezers reveal a detailed view of multimodal diffusion dynamics of the biomacromolecules inside the hierarchically porous structure. Two diffusion constants were parsed from trajectory analyses that were attributed to free diffusion (diffusion constant D = 2.2 μm2/s) and to diffusion within an average confinement length of 210 nm (D = 0.12 μm2/s), consistent with the average macropore size of the coating. Despite its holey nature, the ATO gel network acts as an efficient barrier to the diffusion of the DNA nanostructures, which is strongly indicative of physical interactions between the molecules and the pore nanostructure.
Co-reporter:Sethuramasundaram Pitchiaya, Laurie A. Heinicke, Jun I. Park, Elizabeth L. Cameron, Nils G. Walter
Cell Reports 2017 Volume 19, Issue 3(Volume 19, Issue 3) pp:
Publication Date(Web):18 April 2017
DOI:10.1016/j.celrep.2017.03.075
•iSHiRLoC and CCA quantify miRNA unwinding, turnover, and activity inside cells•miRNA stability and nuclear retention is dependent on Argonaute and targets•miRNA unwinding, strand selection, and cytoplasmic retention are Ago2 dependent•Nuclear miRNAs do not repress cognate targetsRegulation of microRNA (miRNA) localization and stability is critical for their extensive cytoplasmic RNA silencing activity and emerging nuclear functions. Here, we have developed single-molecule fluorescence-based tools to assess the subcellular trafficking, integrity, and activity of miRNAs. We find that seed-matched RNA targets protect miRNAs against degradation and enhance their nuclear retention. While target-stabilized, functional, cytoplasmic miRNAs reside in high-molecular-weight complexes, nuclear miRNAs, as well as cytoplasmic miRNAs targeted by complementary anti-miRNAs, are sequestered stably within significantly lower-molecular-weight complexes and rendered repression incompetent. miRNA stability and activity depend on Argonaute protein abundance, whereas miRNA strand selection, unwinding, and nuclear retention depend on Argonaute identity. Taken together, our results show that miRNA degradation competes with Argonaute loading and target binding to control subcellular miRNA abundance for gene silencing surveillance. Probing single cells for miRNA activity, trafficking, and metabolism promises to facilitate screening for effective miRNA mimics and anti-miRNA drugs.Download high-res image (308KB)Download full-size image
Co-reporter:Soma Dhakal, Matthew R. Adendorff, Minghui Liu, Hao Yan, Mark Bathe and Nils G. Walter
Nanoscale 2016 vol. 8(Issue 5) pp:3125-3137
Publication Date(Web):28 Dec 2015
DOI:10.1039/C5NR07263H
The control of enzymatic reactions using nanoscale DNA devices offers a powerful application of DNA nanotechnology uniquely derived from actuation. However, previous characterization of enzymatic reaction rates using bulk biochemical assays reported suboptimal function of DNA devices such as tweezers. To gain mechanistic insight into this deficiency and to identify design rules to improve their function, here we exploit the synergy of single molecule imaging and computational modeling to characterize the three-dimensional structures and catalytic functions of DNA tweezer-actuated nanoreactors. Our analysis revealed two important deficiencies – incomplete closure upon actuation and conformational heterogeneity. Upon rational redesign of the Holliday junctions located at their hinge and arms, we found that the DNA tweezers could be more completely and uniformly closed. A novel single molecule enzyme assay was developed to demonstrate that our design improvements yield significant, independent enhancements in the fraction of active enzyme nanoreactors and their individual substrate turnover frequencies. The sequence-level design strategies explored here may aid more broadly in improving the performance of DNA-based nanodevices including biological and chemical sensors.
Co-reporter:Krishna C. Suddala; Jiarui Wang; Qian Hou
Journal of the American Chemical Society 2015 Volume 137(Issue 44) pp:14075-14083
Publication Date(Web):October 15, 2015
DOI:10.1021/jacs.5b09740
Bacterial riboswitches couple small-molecule ligand binding to RNA conformational changes that widely regulate gene expression, rendering them potential targets for antibiotic intervention. Despite structural insights, the ligand-mediated folding mechanisms of riboswitches are still poorly understood. Using single-molecule fluorescence resonance energy transfer (smFRET), we have investigated the folding mechanism of an H-type pseudoknotted preQ1 riboswitch in dependence of Mg2+ and three ligands of distinct affinities. We show that, in the absence of Mg2+, both weakly and strongly bound ligands promote pseudoknot docking through an induced-fit mechanism. By contrast, addition of as low as 10 μM Mg2+ generally shifts docking toward conformational selection by stabilizing a folded-like conformation prior to ligand binding. Supporting evidence from transition-state analysis further highlights the particular importance of stacking interactions during induced-fit and of specific hydrogen bonds during conformational selection. Our mechanistic dissection provides unprecedented insights into the intricate synergy between ligand- and Mg2+-mediated RNA folding.
Co-reporter:Kamali N. Sripathi, Pavel Banáš, Kamila Réblová, Jiří Šponer, Michal Otyepka and Nils G. Walter
Physical Chemistry Chemical Physics 2015 vol. 17(Issue 8) pp:5887-5900
Publication Date(Web):21 Jan 2015
DOI:10.1039/C4CP05083E
The hepatitis delta virus (HDV) is the only known human pathogen whose genome contains a catalytic RNA motif (ribozyme). The overall architecture of the HDV ribozyme is that of a double-nested pseudoknot, with two GU pairs flanking the active site. Although extensive studies have shown that mutation of either wobble results in decreased catalytic activity, little work has focused on linking these mutations to specific structural effects on catalytic fitness. Here we use molecular dynamics simulations based on an activated structure to probe the active site dynamics as a result of wobble pair mutations. In both wild-type and mutant ribozymes, the in-line fitness of the active site (as a measure of catalytic proficiency) strongly depends on the presence of a C75(N3H3+)N1(O5′) hydrogen bond, which positions C75 as the general acid for the reaction. Our mutational analyses show that each GU wobble supports catalytically fit conformations in distinct ways; the reverse G25U20 wobble promotes high in-line fitness, high occupancy of the C75(N3H3+)G1(O5′) general-acid hydrogen bond and stabilization of the G1U37 wobble, while the G1U37 wobble acts more locally by stabilizing high in-line fitness and the C75(N3H3+)G1(O5′) hydrogen bond. We also find that stable type I A-minor and P1.1 hydrogen bonding above and below the active site, respectively, prevent local structural disorder from spreading and disrupting global conformation. Taken together, our results define specific, often redundant architectural roles for several structural motifs of the HDV ribozyme active site, expanding the known roles of these motifs within all HDV-like ribozymes and other structured RNAs.
Co-reporter:Rebecca M. Bartke;Elizabeth L. Cameron;Ajitha S. Cristie-David;Thomas C. Custer;Maxwell S. Denies;May Daher;Soma Dhakal;Soumi Ghosh;Laurie A. Heinicke;J. Damon Hoff;Qian Hou;Matthew L. Kahlscheuer;Joshua Karslake;Adam G. Krieger;Jieming Li;Xiang Li;Paul E. Lund;Nguyen N. Vo;Jun Park;Sethuramasundaram Pitchiaya;Victoria Rai;David J. Smith;Krishna C. Suddala;Jiarui Wang;Julia R. Widom
Biopolymers 2015 Volume 103( Issue 5) pp:296-302
Publication Date(Web):
DOI:10.1002/bip.22603
ABSTRACT
Four days after the announcement of the 2014 Nobel Prize in Chemistry for “the development of super-resolved fluorescence microscopy” based on single molecule detection, the Single Molecule Analysis in Real-Time (SMART) Center at the University of Michigan hosted a “Principles of Single Molecule Techniques 2014” course. Through a combination of plenary lectures and an Open House at the SMART Center, the course took a snapshot of a technology with an especially broad and rapidly expanding range of applications in the biomedical and materials sciences. Highlighting the continued rapid emergence of technical and scientific advances, the course underscored just how brightly the future of the single molecule field shines. © 2014 Wiley Periodicals, Inc. Biopolymers 103: 296–302, 2015.
Co-reporter:Leena Mallik, Soma Dhakal, Joseph Nichols, Jacob Mahoney, Anne M. Dosey, Shuoxing Jiang, Roger K. Sunahara, Georgios Skiniotis, and Nils G. Walter
ACS Nano 2015 Volume 9(Issue 7) pp:7133
Publication Date(Web):July 7, 2015
DOI:10.1021/acsnano.5b01841
DNA provides an ideal substrate for the engineering of versatile nanostructures due to its reliable Watson–Crick base pairing and well-characterized conformation. One of the most promising applications of DNA nanostructures arises from the site-directed spatial arrangement with nanometer precision of guest components such as proteins, metal nanoparticles, and small molecules. Two-dimensional DNA origami architectures, in particular, offer a simple design, high yield of assembly, and large surface area for use as a nanoplatform. However, such single-layer DNA origami were recently found to be structurally polymorphous due to their high flexibility, leading to the development of conformationally restrained multilayered origami that lack some of the advantages of the single-layer designs. Here we monitored single-layer DNA origami by transmission electron microscopy (EM) and discovered that their conformational heterogeneity is dramatically reduced in the presence of a low concentration of dimethyl sulfoxide, allowing for an efficient flattening onto the carbon support of an EM grid. We further demonstrated that streptavidin and a biotinylated target protein (cocaine esterase, CocE) can be captured at predesignated sites on these flattened origami while maintaining their functional integrity. Our demonstration that protein assemblies can be constructed with high spatial precision (within ∼2 nm of their predicted position on the platforms) by using strategically flattened single-layer origami paves the way for exploiting well-defined guest molecule assemblies for biochemistry and nanotechnology applications.Keywords: cocaine esterase; DNA nanotechnology; DNA scaffolding; protein−DNA assembly; single-particle electron microscopy;
Co-reporter:Sethuramasundaram Pitchiaya, Laurie A. Heinicke, Thomas C. Custer, and Nils G. Walter
Chemical Reviews 2014 Volume 114(Issue 6) pp:3224
Publication Date(Web):January 8, 2014
DOI:10.1021/cr400496q
Co-reporter:Nils G. Walter and Carlos Bustamante
Chemical Reviews 2014 Volume 114(Issue 6) pp:3069
Publication Date(Web):March 26, 2014
DOI:10.1021/cr500059w
Co-reporter:Julia R. Widom;Soma Dhakal;Laurie A. Heinicke
Archives of Toxicology 2014 Volume 88( Issue 11) pp:1965-1985
Publication Date(Web):2014 November
DOI:10.1007/s00204-014-1357-9
Toxicology is the highly interdisciplinary field studying the adverse effects of chemicals on living organisms. It requires sensitive tools to detect such effects. After their initial implementation during the 1990s, single-molecule fluorescence detection tools were quickly recognized for their potential to contribute greatly to many different areas of scientific inquiry. In the intervening time, technical advances in the field have generated ever-improving spatial and temporal resolution and have enabled the application of single-molecule fluorescence to increasingly complex systems, such as live cells. In this review, we give an overview of the optical components necessary to implement the most common versions of single-molecule fluorescence detection. We then discuss current applications to enzymology and structural studies, systems biology, and nanotechnology, presenting the technical considerations that are unique to each area of study, along with noteworthy recent results. We also highlight future directions that have the potential to revolutionize these areas of study by further exploiting the capabilities of single-molecule fluorescence microscopy.
Co-reporter:Alexander Johnson-Buck, Shuoxing Jiang, Hao Yan, and Nils G. Walter
ACS Nano 2014 Volume 8(Issue 6) pp:5641
Publication Date(Web):May 15, 2014
DOI:10.1021/nn500108k
DNA nanotechnology enables the precise construction of nanoscale devices that mimic aspects of natural biomolecular systems yet exhibit robustly programmable behavior. While many important biological processes involve dynamic interactions between components associated with phospholipid membranes, little progress has been made toward creating synthetic mimics of such interfacial systems. We report the assembly and characterization of cholesterol-labeled DNA origami “barges” capable of reversible association with and lateral diffusion on supported lipid bilayers. Using single-particle fluorescence microscopy, we show that these DNA barges rapidly and stably embed in lipid bilayers and exhibit Brownian diffusion in a manner dependent on both cholesterol labeling and bilayer composition. Tracking of individual barges rapidly generates super-resolution maps of the contiguous regions of a membrane. Addition of appropriate command oligonucleotides enables membrane-associated barges to reversibly exchange fluorescent cargo with bulk solution, dissociate from the membrane, or form oligomers within the membrane, opening up new possibilities for programmable membrane-bound molecular devices.Keywords: diffusion; DNA nanotechnology; DNA origami; lipid bilayer; single-particle tracking
Co-reporter:Alexander Johnson-Buck, Jeanette Nangreave, Shuoxing Jiang, Hao Yan, and Nils G. Walter
Nano Letters 2013 Volume 13(Issue 6) pp:2754-2759
Publication Date(Web):May 23, 2013
DOI:10.1021/nl400976s
We use single-particle fluorescence resonance energy transfer (FRET) to show that organizing oligonucleotide probes into patterned two-dimensional arrays on DNA origami nanopegboards significantly alters the kinetics and thermodynamics of their hybridization with complementary targets in solution. By systematically varying the spacing of probes, we demonstrate that the rate of dissociation of a target is reduced by an order of magnitude in the densest probe arrays. The rate of target binding is reduced less dramatically, but to a greater extent than reported previously for one-dimensional probe arrays. By additionally varying target sequence and buffer composition, we provide evidence for two distinct mechanisms for the markedly slowed dissociation: direct hopping of targets between adjacent sequence-matched probes and nonsequence-specific, salt-bridged, and thus attractive electrostatic interactions with the DNA origami pegboard. This kinetic behavior varies little between individual copies of a given array design and will have significant impact on hybridization measurements and overall performance of DNA nanodevices as well as microarrays.
Co-reporter:Alexander Johnson-Buck, Jeanette Nangreave, Do-Nyun Kim, Mark Bathe, Hao Yan, and Nils G. Walter
Nano Letters 2013 Volume 13(Issue 2) pp:728-733
Publication Date(Web):January 28, 2013
DOI:10.1021/nl304415b
We employ the single-particle fluorescence nanoscopy technique points accumulation for imaging in nanoscale topography (PAINT) using site-specific DNA probes to acquire two-dimensional density maps of specific features patterned on nanoscale DNA origami pegboards. We show that PAINT has a localization accuracy of ∼10 nm that is sufficient to reliably distinguish dense (>104 features μm–2) sub-100 nm patterns of oligonucleotide features. We employ two-color PAINT to follow enzyme-catalyzed modification of features on individual origami and to show that single nanopegboards exhibit stable, spatially heterogeneous probe-binding patterns, or “fingerprints.” Finally, we present experimental and modeling evidence suggesting that these fingerprints may arise from feature spacing variations that locally modulate the probe binding kinetics. Our study highlights the power of fluorescence nanoscopy to perform quality control on individual soft nanodevices that interact with and position reagents in solution.
Co-reporter:Erika N. Cline, Ming-Hsin Li, Seok Ki Choi, Jeffrey F. Herbstman, Neha Kaul, Edgar Meyhöfer, Georgios Skiniotis, James R. Baker, Ronald G. Larson, and Nils G. Walter
Biomacromolecules 2013 Volume 14(Issue 3) pp:
Publication Date(Web):February 7, 2013
DOI:10.1021/bm301719b
Paclitaxel (Taxol) is an anticancer drug that induces mitotic arrest via microtubule hyperstabilization but causes side effects due to its hydrophobicity and cellular promiscuity. The targeted cytotoxicity of hydrophilic paclitaxel-conjugated polyamidoamine (PAMAM) dendrimers has been demonstrated in cultured cancer cells. Mechanisms of action responsible for this cytotoxicity are unknown, that is, whether the cytotoxicity is due to paclitaxel stabilization of microtubules, as is whether paclitaxel is released intracellularly from the dendrimer. To determine whether the conjugated paclitaxel can bind microtubules, we used a combination of ensemble and single microtubule imaging techniques in vitro. We demonstrate that these conjugates adversely affect microtubules by (1) promoting the polymerization and stabilization of microtubules in a paclitaxel-dependent manner, and (2) bundling preformed microtubules in a paclitaxel-independent manner, potentially due to protonation of tertiary amines in the dendrimer interior. Our results provide mechanistic insights into the cytotoxicity of paclitaxel-conjugated PAMAM dendrimers and uncover unexpected risks of using such conjugates therapeutically.
Co-reporter:Matthew S. Marek, Alexander Johnson-Buck and Nils G. Walter
Physical Chemistry Chemical Physics 2011 vol. 13(Issue 24) pp:11524-11537
Publication Date(Web):20 May 2011
DOI:10.1039/C1CP20576E
E Unus pluribum, or “Of One, Many”, may be at the root of decoding the RNA sequence-structure–function relationship. RNAs embody the large majority of genes in higher eukaryotes and fold in a sequence-directed fashion into three-dimensional structures that perform functions conserved across all cellular life forms, ranging from regulating to executing gene expression. While it is the most important determinant of the RNA structure, the nucleotide sequence is generally not sufficient to specify a unique set of secondary and tertiary interactions due to the highly frustrated nature of RNA folding. This frustration results in folding heterogeneity, a common phenomenon wherein a chemically homogeneous population of RNA molecules folds into multiple stable structures. Often, these alternative conformations constitute misfolds, lacking the biological activity of the natively folded RNA. Intriguingly, a number of RNAs have recently been described as capable of adopting multiple distinct conformations that all perform, or contribute to, the same function. Characteristically, these conformations interconvert slowly on the experimental timescale, suggesting that they should be regarded as distinct native states. We discuss how rugged folding free energy landscapes give rise to multiple native states in the Tetrahymena Group I intron ribozyme, hairpin ribozyme, sarcin–ricin loop, ribosome, and an in vitro selected aptamer. We further describe the varying degrees to which folding heterogeneity impacts function in these RNAs, and compare and contrast this impact with that of heterogeneities found in protein folding. Embracing that one sequence can give rise to multiple native folds, we hypothesize that this phenomenon imparts adaptive advantages on any functionally evolving RNA quasispecies.
Co-reporter:Nils G. Walter
Biopolymers 2011 Volume 95( Issue 2) pp:
Publication Date(Web):
DOI:10.1002/bip.21569
No abstract is available for this article.
Co-reporter:Meredith Newby Spano
Biopolymers 2011 Volume 95( Issue 10) pp:653-668
Publication Date(Web):
DOI:10.1002/bip.21626
Abstract
Helix (H)27 of 16S ribosomal (r)RNA from Escherichia coli was dubbed the “switch helix” when mutagenesis suggested that two alternative base pair registers may have distinct functional roles in the bacterial ribosome. Although more recent genetic analyses suggest that H27 conformational switching is not required for translation, previous solution studies demonstrated that the isolated E. coli H27 can dynamically convert between the 885 and 888 conformations. Here, we have solved the nuclear magnetic resonance solution structure of a locked 888 conformation. NOE and residual dipolar coupling restraints reveal an architecture that markedly differs from that of the 885 conformation found in crystal structures of the bacterial ribosome. In place of the loop E motif that characterizes the 885 conformer and that the 888 conformer cannot adopt, we find evidence for an asymmetrical A-rich internal loop stabilized by stacking interactions among the unpaired A's. Comparison of the isolated H27 888 solution structure with the 885 crystal structure within the context of the ribosome suggests a difference in overall length of H27 that presents one plausible reason for the absence of H27 conformational switching within the sterically confining ribosome. © 2011 Wiley Periodicals, Inc. Biopolymers 95: 653–668, 2011.
Co-reporter:Mark A. Ditzler, Michal Otyepka, Jiřì Šponer and Nils G. Walter
Accounts of Chemical Research 2010 Volume 43(Issue 1) pp:40
Publication Date(Web):September 16, 2009
DOI:10.1021/ar900093g
Structure and dynamics are both critical to RNA’s vital functions in biology. Numerous techniques can elucidate the structural dynamics of RNA, but computational approaches based on experimental data arguably hold the promise of providing the most detail. In this Account, we highlight areas wherein molecular dynamics (MD) and quantum mechanical (QM) techniques are applied to RNA, particularly in relation to complementary experimental studies. We have expanded on atomic-resolution crystal structures of RNAs in functionally relevant states by applying explicit solvent MD simulations to explore their dynamics and conformational changes on the submicrosecond time scale. MD relies on simplified atomistic, pairwise additive interaction potentials (force fields). Because of limited sampling, due to the finite accessible simulation time scale and the approximated force field, high-quality starting structures are required. Despite their imperfection, we find that currently available force fields empower MD to provide meaningful and predictive information on RNA dynamics around a crystallographically defined energy minimum. The performance of force fields can be estimated by precise QM calculations on small model systems. Such calculations agree reasonably well with the Cornell et al. AMBER force field, particularly for stacking and hydrogen-bonding interactions. A final verification of any force field is accomplished by simulations of complex nucleic acid structures. The performance of the Cornell et al. AMBER force field generally corresponds well with and augments experimental data, but one notable exception could be the capping loops of double-helical stems. In addition, the performance of pairwise additive force fields is obviously unsatisfactory for inclusion of divalent cations, because their interactions lead to major polarization and charge-transfer effects neglected by the force field. Neglect of polarization also limits, albeit to a lesser extent, the description accuracy of other contributions, such as interactions with monovalent ions, conformational flexibility of the anionic sugar−phosphate backbone, hydrogen bonding, and solute polarization by solvent. Still, despite limitations, MD simulations are a valid tool for analyzing the structural dynamics of existing experimental structures. Careful analysis of MD simulations can identify problematic aspects of an experimental RNA structure, unveil structural characteristics masked by experimental constraints, reveal functionally significant stochastic fluctuations, evaluate the structural role of base ionization, and predict structurally and potentially functionally important details of the solvent behavior, including the presence of tightly bound water molecules. Moreover, combining classical MD simulations with QM calculations in hybrid QM/MM approaches helps in the assessment of the plausibility of chemical mechanisms of catalytic RNAs (ribozymes). In contrast, the reliable prediction of structure from sequence information is beyond the applicability of MD tools. The ultimate utility of computational studies in understanding RNA function thus requires that the results are neither blindly accepted nor flatly rejected, but rather considered in the context of all available experimental data, with great care given to assessing limitations through the available starting structures, force field approximations, and sampling limitations. The examples given in this Account showcase how the judicious use of basic MD simulations has already served as a powerful tool to help evaluate the role of structural dynamics in biological function of RNA.
Co-reporter:Jennifer R. W. Furchak, Peilin Yang, Colin Jennings, Nils G. Walter and Robert T. Kennedy
Analytical Chemistry 2008 Volume 80(Issue 21) pp:8195
Publication Date(Web):October 9, 2008
DOI:10.1021/ac801410k
A naturally occurring aptazyme, the glmS ribozyme, is adapted to an assay for glucosamine 6-phosphate, an effector molecule for the aptazyme. In the assay, binding of analyte allosterically activates aptazyme to cleave a fluorescently labeled oligonucleotide substrate. The extent of reaction, and hence analyte concentration, is detected by either fluorescence resonance energy transfer (FRET) or capillary electrophoresis with laser-induced fluorescence (CE-LIF). With FRET, assay signal is the rate of increase in FRET in presence of analyte. With CE-LIF, the assay signal is the peak height of cleavage product formed after a fixed incubation time. The assay has a linear response up to 100 (CE-LIF) or 500 μM (FRET) and detection limit of ∼500 nM for glucosamine 6-phosphate under single-turnover conditions. When substrate is present in excess of the aptazyme, it is possible to amplify the signal by multiple turnovers to achieve a 13-fold improvement in sensitivity and detection limit of 50 nM. Successful signal amplification requires a temperature cycle to alternately dissociate cleaved substrate and allow fresh substrate to bind aptazyme. The results show that aptazymes have potential utility as analytical reagents for quantification of effector molecules.
Co-reporter:Mark A. Ditzler;Elvin A. Alemán;David Rueda
Biopolymers 2007 Volume 87(Issue 5-6) pp:
Publication Date(Web):8 AUG 2007
DOI:10.1002/bip.20819
The ability of RNA to catalyze chemical reactions was first demonstrated 25 years ago with the discovery that group I introns and RNase P function as RNA enzymes (ribozymes). Several additional ribozymes were subsequently identified, most notably the ribosome, followed by intense mechanistic studies. More recently, the introduction of single molecule tools has dissected the kinetic steps of several ribozymes in unprecedented detail and has revealed surprising heterogeneity not evident from ensemble approaches. Still, many fundamental questions of how RNA enzymes work at the molecular level remain unanswered. This review surveys the current status of our understanding of RNA catalysis at the single molecule level and discusses the existing challenges and opportunities in developing suitable assays. © 2007 Wiley Periodicals, Inc. Biopolymers 87: 302–316, 2007.
This article was originally published online as an accepted preprint. The “Published Online” date corresponds to the preprint version. You can request a copy of the preprint by emailing the Biopolymers editorial office at biopolymers@wiley.com
Co-reporter:S. Elizabeth McDowell;Nad'a Špačková;Jiří Šponer
Biopolymers 2007 Volume 85(Issue 2) pp:
Publication Date(Web):1 NOV 2006
DOI:10.1002/bip.20620
RNA molecules are now known to be involved in the processing of genetic information at all levels, taking on a wide variety of central roles in the cell. Understanding how RNA molecules carry out their biological functions will require an understanding of structure and dynamics at the atomistic level, which can be significantly improved by combining computational simulation with experiment. This review provides a critical survey of the state of molecular dynamics (MD) simulations of RNA, including a discussion of important current limitations of the technique and examples of its successful application. Several types of simulations are discussed in detail, including those of structured RNA molecules and their interactions with the surrounding solvent and ions, catalytic RNAs, and RNA–small molecule and RNA–protein complexes. Increased cooperation between theorists and experimentalists will allow expanded judicious use of MD simulations to complement conceptually related single molecule experiments. Such cooperation will open the door to a fundamental understanding of the structure–function relationships in diverse and complex RNA molecules. © 2006 Wiley Periodicals, Inc. Biopolymers 85:169–184, 2007.
This article was originally published online as an accepted preprint. The “Published Online” date corresponds to the preprint version. You can request a copy of the preprint by emailing the Biopolymers editorial office at biopolymers@wiley.com
Co-reporter:Nils G. Walter
Biopolymers 2007 Volume 85(Issue 2) pp:
Publication Date(Web):16 NOV 2006
DOI:10.1002/bip.20633
The behavior of single molecule defines whether a cell lives, dies, or responds to a specific drug treatment. Single molecule microscopies have begun to reveal the number, location, and functionalities of molecules outside and inside living cells. This issue of Biopolymers presents a first set of reviews that aim to highlight the accomplishments and future prospects of single molecule microscopies. © 2006 Wiley Periodicals, Inc. Biopolymers 85:103–105, 2007.
This article was originally published online as an accepted preprint. The “Published Online” date corresponds to the preprint version. You can request a copy of the preprint by emailing the Biopolymers editorial office at biopolymers@wiley.com
Co-reporter:Jana Sefcikova;Maryna V. Krasovska;Nad'a Špačková;Jiří Šponer
Biopolymers 2007 Volume 85(Issue 5-6) pp:
Publication Date(Web):25 JAN 2007
DOI:10.1002/bip.20693
The self-cleaving hepatitis delta virus (HDV) ribozyme is essential for the replication of HDV, a liver disease causing pathogen in humans. The catalytically critical nucleotide C75 of the ribozyme is buttressed by a trefoil turn pivoting around an extruded G76. In all available crystal structures, the conformation of G76 is restricted by stacking with G76 of a neighboring molecule. To test whether this crystal contact introduces a structural perturbation into the catalytic core, we have analyzed ∼200 ns of molecular dynamics (MD) simulations. In the absence of crystal packing, the simulated G76 fluctuates between several conformations, including one wherein G76 establishes a perpendicular base quadruplet in the major groove of the adjacent P1 stem. Second-site mutagenesis experiments suggest that the identity of the nucleotide in position 76 (N76) indeed contributes to the catalytic activity of a trans-acting HDV ribozyme through its capacity for hydrogen bonding with P1. By contrast, in the cis-cleaving genomic ribozyme the functional relevance of N76 is less pronounced and not correlated with the P1 sequence. Terbium(III) footprinting and additional MD show that the activity differences between N76 mutants of this ribozyme are related instead to changes in average conformation and modified cross-correlations in the trefoil turn. © 2007 Wiley Periodicals, Inc. Biopolymers 85: 392–406, 2007.
This article was originally published online as an accepted preprint. The “Published Online” date corresponds to the preprint version. You can request a copy of the preprint by emailing the Biopolymers editorial office at biopolymers@wiley.com
Co-reporter:David Rueda;Gregory Bokinsky;Maria M. Rhodes;Michael J. Rust;Xiaowei Zhuang;
Proceedings of the National Academy of Sciences 2004 101(27) pp:10066-10071
Publication Date(Web):June 24, 2004
DOI:10.1073/pnas.0403575101
The hairpin ribozyme is a minimalist paradigm for studying RNA folding and function. In this enzyme, two domains dock by induced
fit to form a catalytic core that mediates a specific backbone cleavage reaction. Here, we have fully dissected its reversible
reaction pathway, which comprises two structural transitions (docking/undocking) and a chemistry step (cleavage/ligation),
by applying a combination of single-molecule fluorescence resonance energy transfer (FRET) assays, ensemble cleavage assays,
and kinetic simulations. This has allowed us to quantify the effects that modifications of essential functional groups remote
from the site of catalysis have on the individual rate constants. We find that all ribozyme variants show similar fractionations
into effectively noninterchanging molecule subpopulations of distinct undocking rate constants. This leads to heterogeneous
cleavage activity as commonly observed for RNA enzymes. A modification at the domain junction additionally leads to heterogeneous
docking. Surprisingly, most modifications not only affect docking/undocking but also significantly impact the internal chemistry
rate constants over a substantial distance from the site of catalysis. We propose that a network of coupled molecular motions
connects distant parts of the RNA with its reaction site, which suggests a previously undescribed analogy between RNA and
protein enzymes. Our findings also have broad implications for applications such as the action of drugs and ligands distal
to the active site or the engineering of allostery into RNA.
Co-reporter:Gregory Bokinsky;David Rueda;Vinod K. Misra;Maria M. Rhodes;Andrew Gordus;Hazen P. Babcock;Xiaowei Zhuang;
Proceedings of the National Academy of Sciences 2003 100(16) pp:9302-9307
Publication Date(Web):July 17, 2003
DOI:10.1073/pnas.1133280100
How RNA molecules fold into functional structures is a problem of great
significance given the expanding list of essential cellular RNA enzymes and
the increasing number of applications of RNA in biotechnology and medicine. A
critical step toward solving the RNA folding problem is the characterization
of the associated transition states. This is a challenging task in part
because the rugged energy landscape of RNA often leads to the coexistence of
multiple distinct structural transitions. Here, we exploit single-molecule
fluorescence spectroscopy to follow in real time the equilibrium transitions
between conformational states of a model RNA enzyme, the hairpin ribozyme. We
clearly distinguish structural transitions between effectively
noninterchanging sets of unfolded and folded states and characterize key
factors defining the transition state of an elementary folding reaction where
the hairpin ribozyme's two helical domains dock to make several tertiary
contacts. Our single-molecule experiments in conjunction with site-specific
mutations and metal ion titrations show that the two RNA domains are in a
contact or close-to-contact configuration in the transition state even though
the native tertiary contacts are at most partially formed. Such a compact
transition state without well formed tertiary contacts may be a general
property of elementary RNA folding reactions.
Co-reporter:Nils G. Walter;Dinari A. Harris;Miguel J. B. Pereira;David Rueda
Biopolymers 2002 Volume 61(Issue 3) pp:
Publication Date(Web):25 APR 2002
DOI:10.1002/bip.10144
RNA is a ubiquitous biopolymer that performs a multitude of essential cellular functions involving the maintenance, transfer, and processing of genetic information. RNA is unique in that it can carry both genetic information and catalytic function. Its secondary structure domains, which fold stably and independently, assemble hierarchically into modular tertiary structures. Studies of these folding events are key to understanding how catalytic RNAs (ribozymes) are able to position reaction components for site-specific chemistry. We have made use of fluorescence techniques to monitor the rates and free energies of folding of the small hairpin and hepatitis delta virus (HDV) ribozymes, found in satellite RNAs of plant and the human hepatitis B viruses, respectively. In particular, fluorescence resonance energy transfer (FRET) has been employed to monitor global conformational changes, and 2-aminopurine fluorescence quenching to probe for local structural rearrangements. In this review we illuminate what we have learned about the reaction pathways of the hairpin and HDV ribozymes, and how our results have complemented other biochemical and biophysical investigations. The structural transitions observed in these two small catalytic RNAs are likely to be found in many other biological RNAs, and the described fluorescence techniques promise to be broadly applicable. © 2002 Wiley Periodicals, Inc. Biopoly (Nucleic Acid Sci) 61: 224–241, 2002
Co-reporter:Miguel J.B. Pereira, Evgenia N. Nikolova, Shawna L. Hiley, Dominic Jaikaran, ... Nils G. Walter
Journal of Molecular Biology (3 October 2008) Volume 382(Issue 2) pp:496-509
Publication Date(Web):3 October 2008
DOI:10.1016/j.jmb.2008.07.020
Non-coding RNAs of complex tertiary structure are involved in numerous aspects of the replication and processing of genetic information in many organisms; however, an understanding of the complex relationship between their structural dynamics and function is only slowly emerging. The Neurospora Varkud Satellite (VS) ribozyme provides a model system to address this relationship. First, it adopts a tertiary structure assembled from common elements, a kissing loop and two three-way junctions. Second, catalytic activity of the ribozyme is essential for replication of VS RNA in vivo and can be readily assayed in vitro. Here we exploit single molecule FRET to show that the VS ribozyme exhibits previously unobserved dynamic and heterogeneous hierarchical folding into an active structure. Readily reversible kissing loop formation combined with slow cleavage of the upstream substrate helix suggests a model whereby the structural dynamics of the VS ribozyme favor cleavage of the substrate downstream of the ribozyme core instead. This preference is expected to facilitate processing of the multimeric RNA replication intermediate into circular VS RNA, which is the predominant form observed in vivo.
Co-reporter:Renata A. Rawlings, Vishalakshi Krishnan, Nils G. Walter
Journal of Molecular Biology (29 April 2011) Volume 408(Issue 2) pp:262-276
Publication Date(Web):29 April 2011
DOI:10.1016/j.jmb.2011.02.038
RNA interference is a conserved gene regulatory mechanism employed by most eukaryotes as a key component of their innate immune response to viruses and retrotransposons. During viral infection, the RNase-III-type endonuclease Dicer cleaves viral double-stranded RNA into small interfering RNAs (siRNAs) 21–24 nucleotides in length and helps load them into the RNA-induced silencing complex (RISC) to guide the cleavage of complementary viral RNA. As a countermeasure, many viruses have evolved viral RNA silencing suppressors (RSS) that tightly, and presumably quantitatively, bind siRNAs to thwart RNA-interference-mediated degradation. Viral RSS proteins also act across kingdoms as potential immunosuppressors in gene therapeutic applications. Here we report fluorescence quenching and electrophoretic mobility shift assays that probe siRNA binding by the dimeric RSS p19 from Carnation Italian Ringspot Virus, as well as by human Dicer and RISC assembly complexes. We find that the siRNA:p19 interaction is readily reversible, characterized by rapid binding [(1.69 ± 0.07) × 108 M− 1 s− 1] and marked dissociation (koff = 0.062 ± 0.002 s− 1). We also observe that p19 efficiently competes with recombinant Dicer and inhibits the formation of RISC-related assembly complexes found in human cell extract. Computational modeling based on these results provides evidence for the transient formation of a ternary complex between siRNA, human Dicer, and p19. An expanded model of RNA silencing indicates that multiple turnover by reversible binding of siRNAs potentiates the efficiency of the suppressor protein. Our predictive model is expected to be applicable to the dosing of p19 as a silencing suppressor in viral gene therapy.
Co-reporter:Nils G. Walter
Molecular Cell (28 December 2007) Volume 28(Issue 6) pp:923-929
Publication Date(Web):28 December 2007
DOI:10.1016/j.molcel.2007.12.001
Enzymatic catalysis by RNA was discovered 25 years ago, yet mechanistic insights are emerging only slowly. Thought to be metalloenzymes at first, some ribozymes proved more versatile than anticipated when shown to utilize their own functional groups for catalysis. Recent evidence suggests that some may also judiciously place structural water molecules to shuttle protons in acid-base catalyzed reactions.
Co-reporter:Sethuramasundaram Pitchiaya, Vishalakshi Krishnan, Thomas C. Custer, Nils G. Walter
Methods (15 September 2013) Volume 63(Issue 2) pp:188-199
Publication Date(Web):15 September 2013
DOI:10.1016/j.ymeth.2013.05.028
•Method to probe microRNAs in cellulo with single-molecule sensitivity.•Microinjection and HILO microscopy to deliver and image fluorophore labeled miRNAs.•Single particle tracking in live cells to obtain diffusion coefficients.•Fixed-cell single molecule counting by stepwise photobleaching for stoichiometry.•Mobility and assembly changes reveal intracellular miRNA function.Non-coding RNAs (ncRNAs) recently were discovered to outnumber their protein-coding counterparts, yet their diverse functions are still poorly understood. Here we report on a method for the intracellular Single-molecule High-Resolution Localization and Counting (iSHiRLoC) of microRNAs (miRNAs), a conserved, ubiquitous class of regulatory ncRNAs that controls the expression of over 60% of all mammalian protein coding genes post-transcriptionally, by a mechanism shrouded by seemingly contradictory observations. We present protocols to execute single particle tracking (SPT) and single-molecule counting of functional microinjected, fluorophore-labeled miRNAs and thereby extract diffusion coefficients and molecular stoichiometries of micro-ribonucleoprotein (miRNP) complexes from living and fixed cells, respectively. This probing of miRNAs at the single molecule level sheds new light on the intracellular assembly/disassembly of miRNPs, thus beginning to unravel the dynamic nature of this important gene regulatory pathway and facilitating the development of a parsimonious model for their obscured mechanism of action.
Co-reporter:Alexander Johnson-Buck, Nils G. Walter
Methods (15 May 2014) Volume 67(Issue 2) pp:177-184
Publication Date(Web):15 May 2014
DOI:10.1016/j.ymeth.2014.02.032
•We review two methods for measuring hybridization kinetics on DNA nanostructures.•Single-molecule fluorescence microscopy detects unexpected hybridization anomalies.•Single-particle FRET reveals hybridization kinetics dependent on probe density.•DNA-PAINT uncovers variable binding kinetics across nanostructures.DNA nanostructures are finding diverse applications as scaffolds for molecular organization. In general, components such as nucleic acids, proteins, and nanoparticles are attached to addressable DNA nanostructures via hybridization, and there is interest in exploiting hybridization for localized computation on DNA nanostructures. This report details two fluorescence microscopy methods, single-particle fluorescence resonance energy transfer (spFRET) and DNA-PAINT (points accumulation for imaging in nanoscale topography), that have been successfully used to detect anomalies of hybridization reactions on individual DNA nanostructures. We compare and contrast the two techniques, highlighting their respective strengths in studying equilibrium and non-equilibrium hybridization as well as assessing the variability of behaviors within individual nanostructures and across a population of nanostructures.
Co-reporter:Matthew S. Marek, Alexander Johnson-Buck and Nils G. Walter
Physical Chemistry Chemical Physics 2011 - vol. 13(Issue 24) pp:NaN11537-11537
Publication Date(Web):2011/05/20
DOI:10.1039/C1CP20576E
E Unus pluribum, or “Of One, Many”, may be at the root of decoding the RNA sequence-structure–function relationship. RNAs embody the large majority of genes in higher eukaryotes and fold in a sequence-directed fashion into three-dimensional structures that perform functions conserved across all cellular life forms, ranging from regulating to executing gene expression. While it is the most important determinant of the RNA structure, the nucleotide sequence is generally not sufficient to specify a unique set of secondary and tertiary interactions due to the highly frustrated nature of RNA folding. This frustration results in folding heterogeneity, a common phenomenon wherein a chemically homogeneous population of RNA molecules folds into multiple stable structures. Often, these alternative conformations constitute misfolds, lacking the biological activity of the natively folded RNA. Intriguingly, a number of RNAs have recently been described as capable of adopting multiple distinct conformations that all perform, or contribute to, the same function. Characteristically, these conformations interconvert slowly on the experimental timescale, suggesting that they should be regarded as distinct native states. We discuss how rugged folding free energy landscapes give rise to multiple native states in the Tetrahymena Group I intron ribozyme, hairpin ribozyme, sarcin–ricin loop, ribosome, and an in vitro selected aptamer. We further describe the varying degrees to which folding heterogeneity impacts function in these RNAs, and compare and contrast this impact with that of heterogeneities found in protein folding. Embracing that one sequence can give rise to multiple native folds, we hypothesize that this phenomenon imparts adaptive advantages on any functionally evolving RNA quasispecies.
Co-reporter:Kamali N. Sripathi, Pavel Banáš, Kamila Réblová, Jiří Šponer, Michal Otyepka and Nils G. Walter
Physical Chemistry Chemical Physics 2015 - vol. 17(Issue 8) pp:NaN5900-5900
Publication Date(Web):2015/01/21
DOI:10.1039/C4CP05083E
The hepatitis delta virus (HDV) is the only known human pathogen whose genome contains a catalytic RNA motif (ribozyme). The overall architecture of the HDV ribozyme is that of a double-nested pseudoknot, with two GU pairs flanking the active site. Although extensive studies have shown that mutation of either wobble results in decreased catalytic activity, little work has focused on linking these mutations to specific structural effects on catalytic fitness. Here we use molecular dynamics simulations based on an activated structure to probe the active site dynamics as a result of wobble pair mutations. In both wild-type and mutant ribozymes, the in-line fitness of the active site (as a measure of catalytic proficiency) strongly depends on the presence of a C75(N3H3+)N1(O5′) hydrogen bond, which positions C75 as the general acid for the reaction. Our mutational analyses show that each GU wobble supports catalytically fit conformations in distinct ways; the reverse G25U20 wobble promotes high in-line fitness, high occupancy of the C75(N3H3+)G1(O5′) general-acid hydrogen bond and stabilization of the G1U37 wobble, while the G1U37 wobble acts more locally by stabilizing high in-line fitness and the C75(N3H3+)G1(O5′) hydrogen bond. We also find that stable type I A-minor and P1.1 hydrogen bonding above and below the active site, respectively, prevent local structural disorder from spreading and disrupting global conformation. Taken together, our results define specific, often redundant architectural roles for several structural motifs of the HDV ribozyme active site, expanding the known roles of these motifs within all HDV-like ribozymes and other structured RNAs.
.jpg)


















![2-amino-4,7-dihydro-4-oxo-3H-Pyrrolo[2,3-d]pyrimidine-5-carbonitrile](http://img.cochemist.com/ccimg/69300/69205-79-4.png)
![2-amino-4,7-dihydro-4-oxo-3H-Pyrrolo[2,3-d]pyrimidine-5-carbonitrile](http://img.cochemist.com/ccimg/69300/69205-79-4_b.png)




![Methyl (3s,4r)-3-benzoyloxy-8-methyl-8-azabicyclo[3.2.1]octane-4-carboxylate](http://img.cochemist.com/ccimg/100/50-36-2.png)
![Methyl (3s,4r)-3-benzoyloxy-8-methyl-8-azabicyclo[3.2.1]octane-4-carboxylate](http://img.cochemist.com/ccimg/100/50-36-2_b.png)














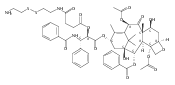
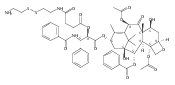
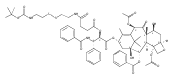
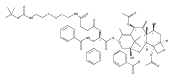


















![2-amino-5-(aminomethyl)-3,7-dihydro-4H-Pyrrolo[2,3-d]pyrimidin-4-one](http://img.cochemist.com/ccimg/69300/69251-45-2.png)
![2-amino-5-(aminomethyl)-3,7-dihydro-4H-Pyrrolo[2,3-d]pyrimidin-4-one](http://img.cochemist.com/ccimg/69300/69251-45-2_b.png)






![3H-Indolium, 2-[5-[1-[6-[(2,5-dioxo-1-pyrrolidinyl)oxy]-6-oxohexyl]-1,3-dihydro-3,3-dimethyl-5-sulfo-2H-indol-2-ylidene]-1,3-pentadien-1-yl]-1-ethyl-3,3-](/data/chemimg/105700/146368-14-1.png)
![3H-Indolium, 2-[5-[1-[6-[(2,5-dioxo-1-pyrrolidinyl)oxy]-6-oxohexyl]-1,3-dihydro-3,3-dimethyl-5-sulfo-2H-indol-2-ylidene]-1,3-pentadien-1-yl]-1-ethyl-3,3-](/data/chemimg/105700/146368-14-1_b.png)