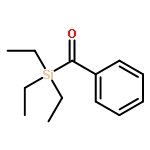
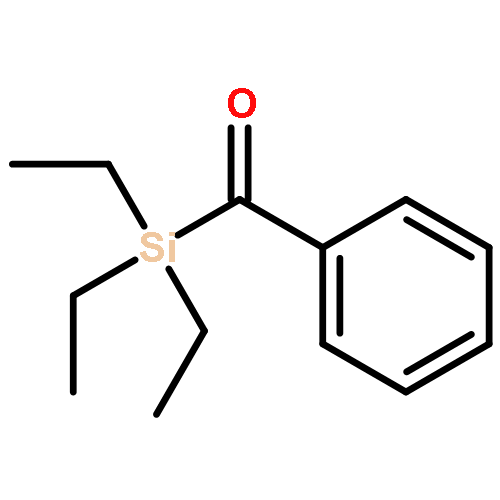
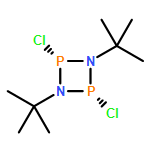
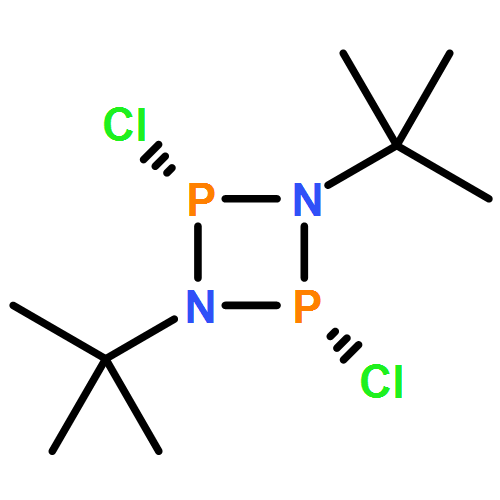
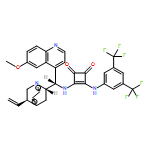
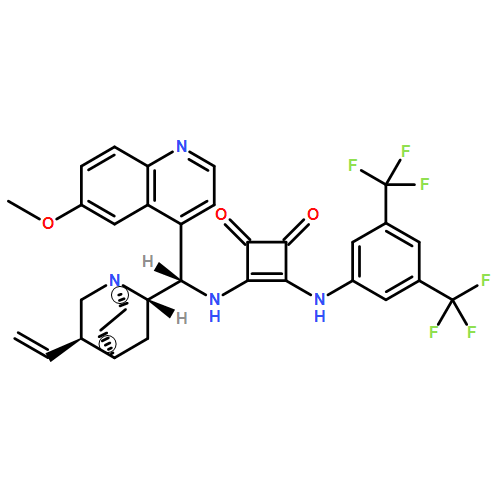
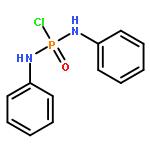
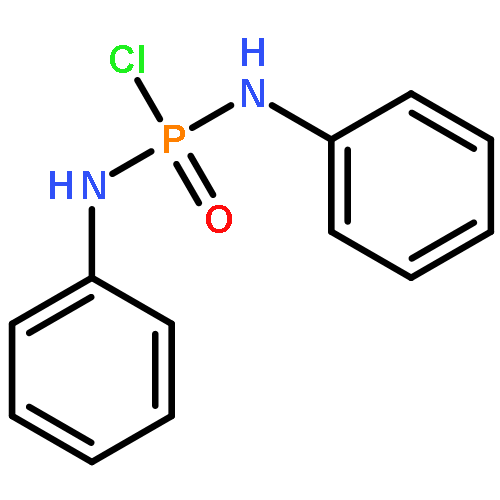
Modular cyclodiphosph(V)azanes are synthesised and their affinity for chloride and actetate anions were compared to those of a bisaryl urea derivative (1). The diamidocyclodiphosph(V)azanes cis-[{ArNHP(O)(μ-tBu)}2] [Ar=Ph (2) and Ar=m-(CF3)2Ph (3)] were synthesised by reaction of [{ClP(μ-NtBu)}2] (4) with the respective anilines and subsequent oxidation with H2O2. Phosphazanes 2 and 3 were obtained as the cis isomers and were characterised by multinuclear NMR spectroscopy, FTIR spectroscopy, HRMS and single-crystal X-ray diffraction. The cyclodiphosphazanes 2 and 3 readily co-crystallise with donor solvents such as MeOH, EtOH and DMSO through bidentate hydrogen bonding, as shown in the X-ray analyses. Cyclodiphosphazane 3 showed a remarkably high affinity (log[K]=5.42) for chloride compared with the bisaryl urea derivative 1 (log[K]=4.25). The affinities for acetate (AcO−) are in the same range (3: log[K]=6.72, 1: log[K]=6.91). Cyclodiphosphazane 2, which does not contain CF3 groups, exhibits weaker binding to chloride (log[K]=3.95) and acetate (log[K]=4.49). DFT computations and X-ray analyses indicate that a squaramide-like hydrogen-bond directionality and CαH interactions account for the efficiency of 3 as an anion receptor. The CαH groups stabilise the Z,Z-3 conformation, which is necessary for bidentate hydrogen bonding, as well as coordinating with the anion.
A series of bulky, modular, monodentate, fenchol-based phosphites has been employed in an intramolecular palladium-catalyzed alkyl-aryl cross-coupling reaction. This enantioselective α-arylation of N-(2-bromophenyl)-N-methyl-2-phenylpropanamide is accomplished with [Pd(C3H5)(BIFOP-X)(Cl)] as precatalysts, which are based on biphenyl-2,2′-bisfenchol phosphites (BIFOP-X, X=F, Cl, Br, etc.). The phosphorus fluoride BIFOP-F gives the highest enantioselectivity and good yields (64% ee, 88%). Lower selectivities and yields are found for BIFOP halides with heavier halogens (Cl: 74%, 47% ee, Br: 63%, 20% ee). NMR studies on catalyst complexes reveal two equilibrating diastereomeric complexes in equal proportions. In all cases, the phosphorus-halogen moiety remains intact, pointing to its remarkable stability, even in the presence of nucleophiles. The increasing enantioselectivity of the catalysts with the phosphorus halide ligands correlates with the rising electronegativity of the halide (bromine<chlorine<fluorine), as can be rationalized from structural parameters and DFT computations.
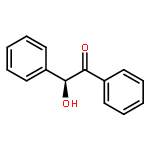
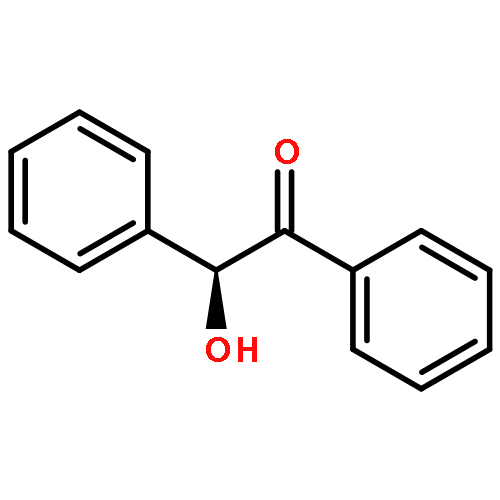
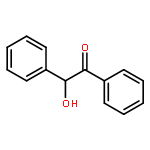
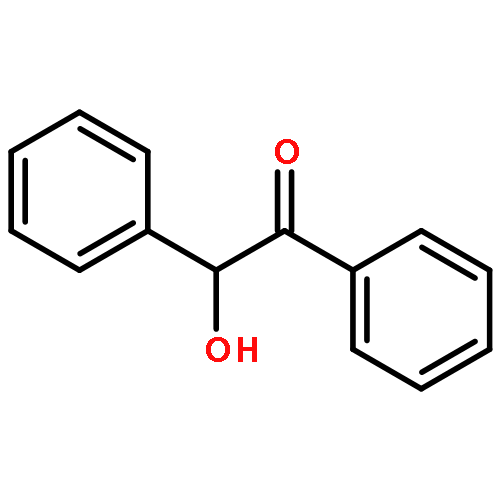
Two new classes of terpenol-based lithium phosphonates, i.e. phenylfenchyl phosphonates and 2,2′-biphenyldiylbis(terpenyl) phosphonates, are employed as umpolung catalysts for the enantioselective cross benzoin coupling. The structural characteristics of the phosphonates were investigated by means of X-ray, 31P-NMR analyses as well as DFT computations. The chiral lithium phosphonates were found to catalyze the cross benzoin coupling with enantioselectivities up to 54 % ee. For the phenylfenchyl phosphonates, the catalyst reactivities highly depend on the substituents in benzylic position. Modification of benzylic >CH2 to >C(CF3)2 significantly increases the yield from 19 to 92 %. For phenylfenchyl phosphonate pre-catalysts, substitution of the benzylic >CH2 group by >C(CF3)2 changes the sense of enantioselectivity from S- to the R-configured benzoin product.
New enantiopure pyridyl alcohols are efficiently accessible through few synthetic steps from commercially available terpenes, that is, (+)-fenchone, (−)-menthone and (−)-verbenone as well as 2,6-diphenylpyridine. These chelating pyridyl alcohols exhibit flexible pyridyl–phenylene axes, which give rise to P and M conformers. Alkylzincation of the hydroxy groups eliminates equilibria of the conformers and generates alkylzinc complexes with adjusted biaryl axes, as it is demonstrated by NMR studies. These alkylzinc catalysts perform well in the addition of dimethylzinc or diethylzinc to benzaldehyde with yields up to 99 % and ee’s up to 95 %. The adjusted pyridylphenylene conformations in the ligands now control enantioselectivities of the catalysts, which were also analysed by computations at the DFT level.
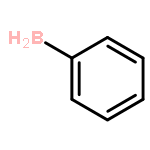
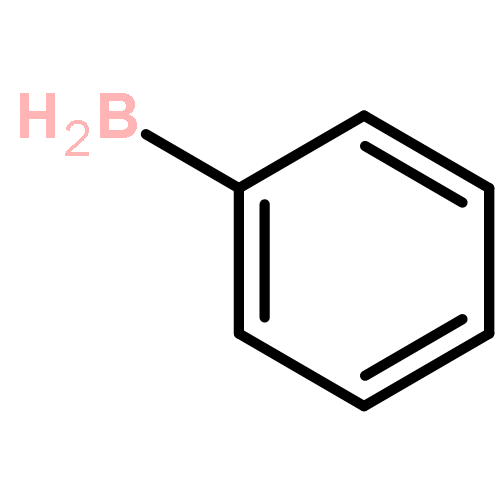
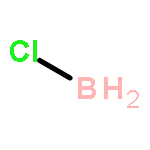
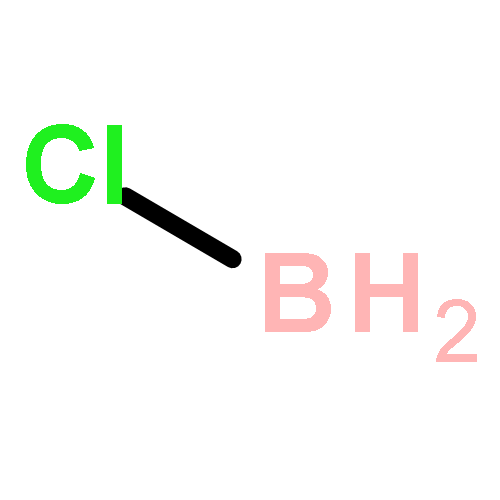
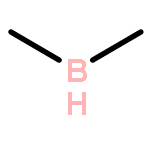
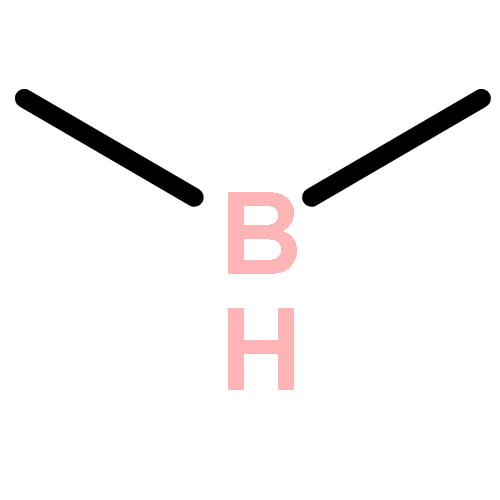
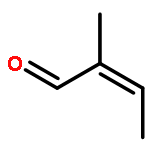
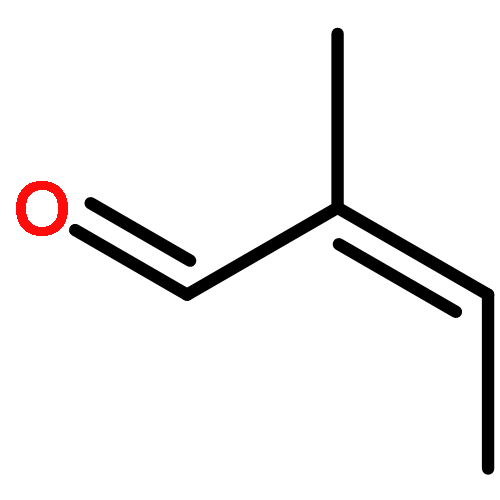
The CH functionalization of methane by means of direct CH borations with BH3 or MeBH2 is compared computationally (using the B3LYP/6-311++G** method) to CH lithiations with LiH or LiMe as well as to other analogue C–metal (Be, Na, Mg, Al) formations. For the borations only, this internal electrophilic substitution at carbon (SEi) relies more on the electrophilicity of boron than on the basicity of the internal base Y, that is, H or Me. Such direct borations of methane are more favored for dehydrogenations than for dehydrocarbonations. Due to decreased electrophilicity, substituents at boron disfavor such borations. Hence, the BH2 group appears to be most efficient for CH functionalizations by means of direct hydrocarbon borations.
Phenyl fenchol forms a 3:1 aggregate with n-butyllithium (3-BuLi), showing unique lithium–HC agostic interactions both in toluene solution (1H,7Li-HOESY) and in the solid state (X-ray analysis). Although methoxy–lithium coordination is characteristic for many mixed aggregates of anisyl fencholates with n-butyllithium, endo-methyl coordination to lithium ions compensates for the missing methoxy groups in 3-BuLi. This gives rise to a different orientation of the fenchane moiety, encapsulating and chirally modifying the butylide unit.
Modular fenchyl phosphinites (FENOPs) containing different aryl units—phenyl (1), 2-anisyl (2), or 2-pyridyl (3)—are efficiently accessible from (−)-fenchone. For comparison of the influence of the different aryl units on enantioselectivities and reactivities, these FENOPs were employed in Pd-catalyzed allylic alkylations. The strongly chelating character of P,N-bidentate 3 is apparent from X-ray structures with PdCl2 ([Pd(3)Cl2]), and with allyl–Pd units in ([Pd(3)(η1-allyl)] and [Pd(3)(η3-allyl)]). FENOP3 gives rise to a PdL* catalyst of moderate enantioselectivity (42 % ee, R product). Surprisingly, higher enantioselectivities are found for the hemilabile, monodentate FENOPs 1 (83 % ee, S enantiomer) and 2 (69 % ee, S enantiomer). Only small amounts of 1 or 2 generate selective PdL* catalysts, while complete abolition of enantioselectivity appears with unselective PdL*2 species with higher FENOP concentrations in the cases of 1 or 2. Computational transition structure analyses reveal steric and electronic origins of enantioselectivities. The nucleophile is electronically guided trans to phosphorus. endo-Allyl arrangements are favored over exo-allyl orientations for 1 and 2 due to Pd–π–pyridyl interactions with short “side-on” Pd-aryl interactions. More remote “edge-on” Pd–π–aryl interactions in 3 with Pd-N(lp) coordination favor endo-allyl units slightly more and explain the switch of enantioselectivity from 1 (S) and 2 (S) to 3 (R).
Modulare Fenchylphosphinite (FENOPs) mit unterschiedlichen Arylgruppen—d.h. Phenyl (1), 2-Anisyl (2) oder 2-Pyridyl (3)—sind effizient aus (−)-Fenchon zugänglich. Zum Vergleich unterschiedlicher Arylgruppen hinsichtlich der erzielten Enantioselektivitäten und Reaktivitäten, wurden diese FENOPs in Pd-katalysierten allylischen Alkylierungen eingesetzt. Der stark chelatisierende Charakter des bidentaten P,N-Liganden 3 wird durch Röntgenstrukturen mit PdCl2([Pd(3)Cl2]), sowie mit Allyleinheiten ([Pd(3)(η1-allyl)] und [Pd(3)(η3-allyl)]) deutlich. Der bidentate P,N-Ligand 3 führt zu einem PdL* Katalysator mit moderater Enantioselektivität (42 % ee, R Produkt). Überraschenderweise werden für die hemilabilen FENOP Liganden 1 (83 % ee, S Enantiomer) und 2 (69 % ee, S Enantiomer) höhere Enantioselektivitäten des anderen Enantiomers gefunden. Nur kleinere Mengen von 1 oder 2 bilden selektive PdL* Katalysatoren, während mit höheren FENOP Konzentrationen unselektive PdL*2Spezies die Enantioselektivitäten zusammenbrechen lassen. Theoretische Berechnungen an Übergangszuständen offenbaren sterische und elektronische Ursprünge der Enantioselektivitäten. Das Nukleophil wird elektronisch kontrolliert trans zum P-Atom geleitet. Für 1 und 2 sind endo- gegenüber exo-Allyl Anordnungen wegen Pd–π–Pyridyl Wechselwirkungen mit kurzen “side-on” Pd-Aryl Kontakten bevorzugt. In 3 begünstigen mit ihrem größerem Abstand “edge-on” Pd–π–Aryl Wechselwirkungen durch Pd-N(lp) Koordination die endo-Allyl Anordnungen etwas mehr und führen so zum Wechsel des bevorzugten Enantiomers von 1 (S) und 2 (S) zu 3 (R).
Scalemic mixtures of chiral anisyl fenchols with different ortho-substituents (X) in the anisyl moieties [X=H (1), Me (2), SiMe3 (3) and tBu (4)] are employed as pre-catalysts in enantioselective additions of diethylzinc to benzaldehyde. While a remarkable asymmetric depletion is apparent for X=H and Me, a linear relationship between the enantiomeric purity of the chiral source and the product 1-phenylpropanol is observed for X=SiMe3 and tBu. X-ray single crystal analyses show that racemic methylzinc fencholates obtained from 1 (X=H) and 2 (X=Me) yield homochiral dimeric complexes, while for 3 (X=SiMe3) and 4 (X=tBu) the heterochiral dimeric alkylzinc structures are formed. The enantiopure fenchols 1–4 all yield homochiral dimeric methylzinc complexes. Computed relative energies of homo- and heterochiral fencholate dimers with X=H and Me reveal an intrinsic preference for the formation of the homochiral dimers, consistent with the observed negative NLE. In contrast, similar stabilities are computed for homo- and heterochiral complexes of ligands 3 (X=SiMe3) and 4 (X=tBu), in agreement with the absence of a nonlinear effect for bulky ortho-substituents.
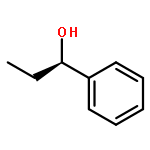
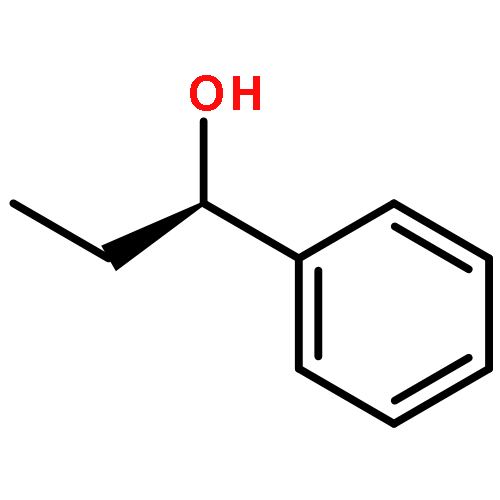
![2H-1-BENZOPYRAN-2-ONE, 4-HYDROXY-3-[(1R)-3-OXOCYCLOHEXYL]-](http://img.cochemist.com/ccimg/547800/547743-10-2.png)
![2H-1-BENZOPYRAN-2-ONE, 4-HYDROXY-3-[(1R)-3-OXOCYCLOHEXYL]-](http://img.cochemist.com/ccimg/547800/547743-10-2_b.png)
![Bicyclo[2.2.1]heptan-2-ol, 1,3,3-trimethyl-2-(2-pyridinyl)-, (1R,2R,4S)-](http://img.cochemist.com/ccimg/191500/191402-03-6.png)
![Bicyclo[2.2.1]heptan-2-ol, 1,3,3-trimethyl-2-(2-pyridinyl)-, (1R,2R,4S)-](http://img.cochemist.com/ccimg/191500/191402-03-6_b.png)
![Propanedioic acid, [(1R)-1-phenyl-2-propenyl]-, dimethyl ester](http://img.cochemist.com/ccimg/164800/164713-27-3.png)
![Propanedioic acid, [(1R)-1-phenyl-2-propenyl]-, dimethyl ester](http://img.cochemist.com/ccimg/164800/164713-27-3_b.png)
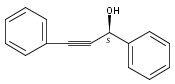
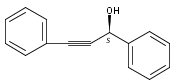
![Propanedioic acid, [(1S)-1-phenyl-2-propenyl]-, dimethyl ester](http://img.cochemist.com/ccimg/129200/129101-14-0.png)
![Propanedioic acid, [(1S)-1-phenyl-2-propenyl]-, dimethyl ester](http://img.cochemist.com/ccimg/129200/129101-14-0_b.png)
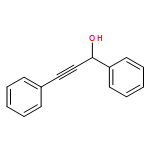
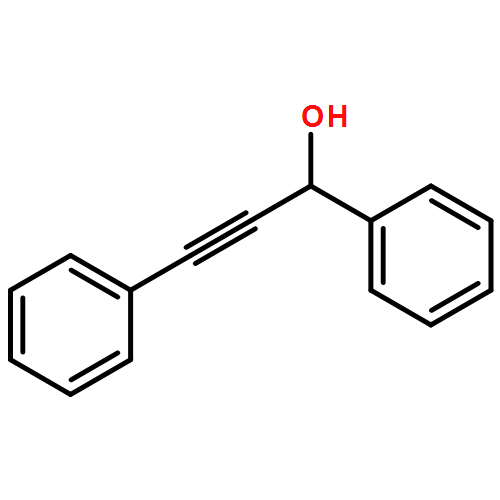
![Propanedioic acid, [(1R,2E)-1,3-diphenyl-2-propenyl]-, dimethyl ester](http://img.cochemist.com/ccimg/96500/96482-67-6.png)
![Propanedioic acid, [(1R,2E)-1,3-diphenyl-2-propenyl]-, dimethyl ester](http://img.cochemist.com/ccimg/96500/96482-67-6_b.png)
![Propanedioic acid, [(1S,2E)-1,3-diphenyl-2-propenyl]-, dimethyl ester](http://img.cochemist.com/ccimg/96500/96482-64-3.png)
![Propanedioic acid, [(1S,2E)-1,3-diphenyl-2-propenyl]-, dimethyl ester](http://img.cochemist.com/ccimg/96500/96482-64-3_b.png)

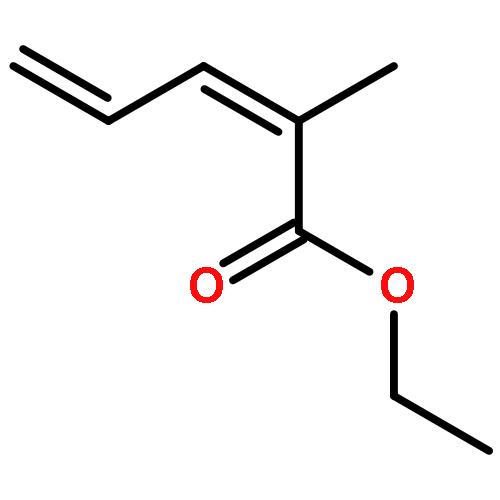
![Benzene, [(1E)-3-methyl-1,3-butadienyl]-](http://img.cochemist.com/ccimg/68100/68036-69-1.png)
![Benzene, [(1E)-3-methyl-1,3-butadienyl]-](http://img.cochemist.com/ccimg/68100/68036-69-1_b.png)

![Ethanone, 1,2-diphenyl-2-[(trimethylsilyl)oxy]-](http://img.cochemist.com/ccimg/26300/26205-39-0.png)
![Ethanone, 1,2-diphenyl-2-[(trimethylsilyl)oxy]-](http://img.cochemist.com/ccimg/26300/26205-39-0_b.png)
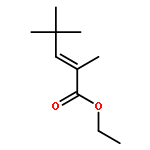
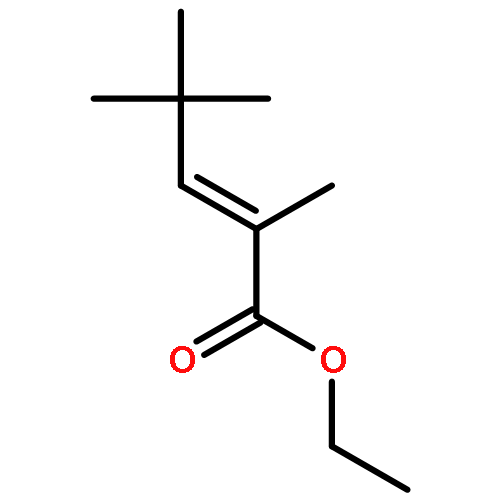
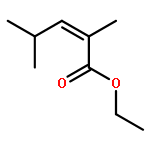
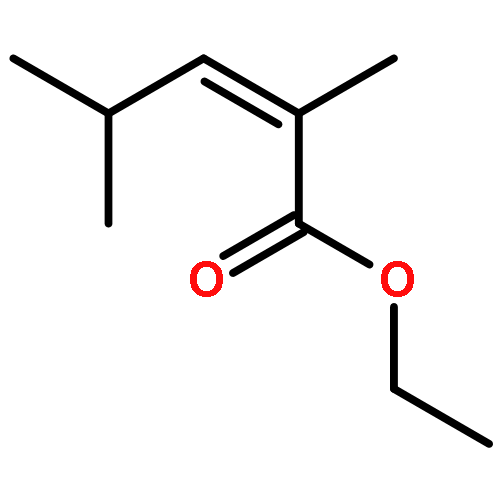
![Di-μ-chlorobis[(1,2,3-η)-1-phenyl-2-propenyl]dipalladium(Ⅱ)](http://img.cochemist.com/ccimg/12200/12131-44-1.png)
![Di-μ-chlorobis[(1,2,3-η)-1-phenyl-2-propenyl]dipalladium(Ⅱ)](http://img.cochemist.com/ccimg/12200/12131-44-1_b.png)
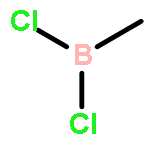
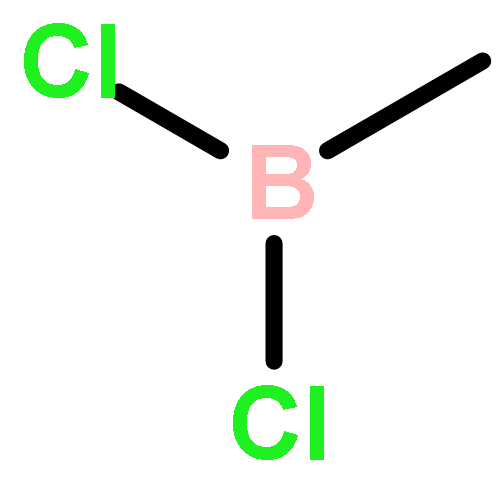
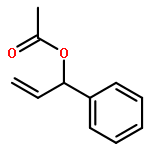
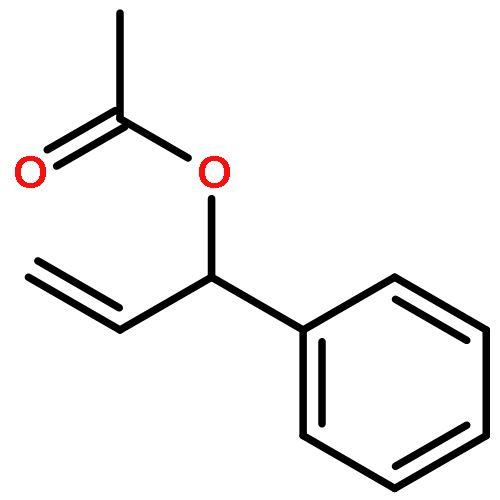
![2H-1-Benzopyran-2-one,4-hydroxy-3-[(1R)-3-oxo-1-phenylbutyl]-](http://img.cochemist.com/ccimg/5600/5543-58-8.png)
![2H-1-Benzopyran-2-one,4-hydroxy-3-[(1R)-3-oxo-1-phenylbutyl]-](http://img.cochemist.com/ccimg/5600/5543-58-8_b.png)
![1,3-BIS[3,5-BIS(TRIFLUOROMETHYL)PHENYL]UREA](http://img.cochemist.com/ccimg/3900/3824-74-6.png)
![1,3-BIS[3,5-BIS(TRIFLUOROMETHYL)PHENYL]UREA](http://img.cochemist.com/ccimg/3900/3824-74-6_b.png)
![N,N-DIMETHYL-N-{4-[2-NITROVINYL]PHENYL}AMINE](http://img.cochemist.com/ccimg/2700/2604-08-2.png)
![N,N-DIMETHYL-N-{4-[2-NITROVINYL]PHENYL}AMINE](http://img.cochemist.com/ccimg/2700/2604-08-2_b.png)
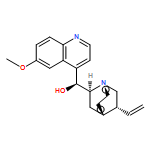
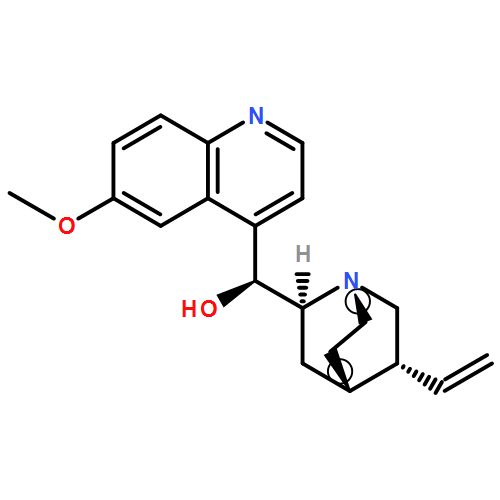


![BICYCLO[2.2.1]HEPTAN-2-OL, 1,3,3-TRIMETHYL-2-PHENYL-, (1R,2R,4S)-](http://img.cochemist.com/ccimg/161300/161276-73-9.png)
![BICYCLO[2.2.1]HEPTAN-2-OL, 1,3,3-TRIMETHYL-2-PHENYL-, (1R,2R,4S)-](http://img.cochemist.com/ccimg/161300/161276-73-9_b.png)