Co-reporter:Andrea Lauer, David E. Fast, Jan Steinkoenig, Anne-Marie Kelterer, Georg Gescheidt, and Christopher Barner-Kowollik
ACS Macro Letters September 19, 2017 Volume 6(Issue 9) pp:952-952
Publication Date(Web):August 16, 2017
DOI:10.1021/acsmacrolett.7b00499
Herein, we report the unique—and first time—wavelength-dependent investigation with strictly monochromatic light of 305–405 nm wavelength into the stability of photoinitiator-derived chain termini of poly(methyl methacrylate) using a tunable laser system fused with pulsed-laser irradiation and size exclusion chromatography hyphenated to high-resolution electrospray mass spectrometry (PLI-SEC-ESI-MS). We assess several substitution patterns of methyl groups on the common benzoyl-type radical fragment. Critically, methyl substitution in the 2- and 6-positions of the benzoyl moiety, i.e., in both ortho-positions, resulted in stable chain ends up to approximately 350 nm. The stability can be attributed to a blue-shift of the n−π* transitions (relevant for the end group reactivity) as predicted by earlier density functional theory (DFT) calculations on model species. In sharp contrast, our experiments show a far reduced stability of the end groups commencing from 400 nm onwards, when the dual ortho-methyl substitution in the benzoyl fragment is missing. Thus, we demonstrate that the substitution pattern on the phenyl ring of the benzoyl group dictates the chain end stability as a function of wavelength in excellent agreement with the quantum chemical predictions. Our study thus provides critical insights into selecting suitable photoinitiation systems for specific wavelength regimes.
Co-reporter:Kilian N. R. Wuest, Hongxu Lu, Donald S. Thomas, Anja S. Goldmann, Martina H. Stenzel, and Christopher Barner-Kowollik
ACS Macro Letters October 17, 2017 Volume 6(Issue 10) pp:1168-1168
Publication Date(Web):October 4, 2017
DOI:10.1021/acsmacrolett.7b00659
We introduce the light-induced collapse of single glycopolymer chains in water generating fluorescent glyco single-chain nanoparticles (SCNPs) and their subsequent functionalization onto nanodiamonds. The glycopolymer precursors are prepared by polymerizing an acetylated mannose-based methacrylate monomer followed by a deprotection and postpolymerization functionalization step, introducing profluorescent photoactive tetrazole groups and furan-protected maleimide moieties. Subsequent UV irradiation in highly diluted aqueous solution triggers intramolecular tetrazole-mediated cycloadditions, yielding glyco SCNPs featuring fluorescence as well as lectin binding properties. The obtained SCNPs are coated onto nanodiamonds by adsorption, and the obtained hybrid nanoparticles are in depth characterized in terms of size, functionality, and bioactivity. Different coating densities are achieved by altering the SCNP concentration. The prepared nanoparticles are nontoxic in mouse RAW 264.7 macrophages. Furthermore, the fluorescence of the SCNPs can be exploited to image the SCNP-coated nanodiamonds in macrophage cells via confocal fluorescence microscopy.
Co-reporter:Philipp Jöckle, Caroline Schweigert, Iris Lamparth, Norbert Moszner, Andreas-Neil Unterreiner, and Christopher Barner-Kowollik
Macromolecules November 28, 2017 Volume 50(Issue 22) pp:8894-8894
Publication Date(Web):November 9, 2017
DOI:10.1021/acs.macromol.7b01721
Five para-substituted monoacyltrimethylgermane derivatives, i.e., p-fluorobenzoyltrimethylgermane (pFBG, λmax = 405 nm), p-methoxybenzoyltrimethylgermane (pMBG, λmax = 397 nm), benzoyltrimethylgermane (pHBG, λmax = 409 nm), p-cyanobenzoyltrimethylgermane (pCBG, λmax = 425 nm), and p-nitrobenzoyltrimethylgermane (pNBG, λmax = 429 nm) are investigated via a combination of pulsed laser polymerization with subsequent electrospray ionization and mass spectrometry (PLP-ESI-MS) as well as femtosecond transient absorption spectroscopy. The relative initiation efficiencies of the initiating benzoyl radical fragments of pFBG, pMBG, and pHBG are determined using PLP-ESI-MS. The para-substituted derivatives with the electron-donating groups, pFBG and pMBG, display a factor 1.5 and 1.3, respectively, superior overall initiation efficiency compared to the unsubstituted pHBG. In contrast, the derivatives pCBG and pNBG carrying electron-withdrawing groups display only weak initiation behavior at a factor 4 higher total energy of ∼112 J (∼28 J for typical PLP experiments with pMBG, pFBG, and pHBG at ∼320 J and 90 000 pulses). The differences in the initiation efficiencies are representative for two classes of monoacyltrimethylgermane initiators, i.e., efficient initiators and weak initiators, each distinct in their specific radical cleavage mechanism. The efficient initiators pMBG, pFBG, and pHBG show an ultrafast intersystem crossing within 2–4 ps after pulse irradiation and subsequent formation of benzoyl and trimethylgermyl radical fragments. In contrast, the weak initiators pCBG and pNBG relax to the ground state after photoexcitation via a dominating ultrafast internal conversion (IC) within 13 and 2 ps, respectively, disallowing effective initiation under typical PLP conditions (∼320 J/pulse with 90 000 pulses resulting in ∼28 J total energy per sample). pCBG features weak initiation behavior additionally forming methyl and p-cyanobenzoyldimethylgermyl radicals at a factor 4 higher total energy of ∼112 J. Consistent with a considerably faster IC relaxation, pNBG features a factor 10 weaker monomer conversion than pCBG.
Co-reporter:Bernhard V. K. J. Schmidt, Dennis Kugele, Jonas von Irmer, Jan Steinkoenig, Hatice Mutlu, Christian Rüttiger, Craig J. Hawker, Markus Gallei, and Christopher Barner-Kowollik
Macromolecules March 28, 2017 Volume 50(Issue 6) pp:2375-2375
Publication Date(Web):March 14, 2017
DOI:10.1021/acs.macromol.7b00165
We introduce the formation of a dual responsive supramolecular star polymer system, displaying gated self-assembly behavior. The redox- and thermoresponsive star polymers are based on a 6-fold β-CD functionalized core molecule and RAFT-derived ferrocene end functionalized poly(N,N-dimethylacrylamide) (PDMA) and poly(N,N-diethylacrylamide) (PDEA) linear polymers. Complex formation is analyzed via various methods including dynamic light scattering (DLS). Chemical redox triggers, namely NaOCl and ascorbic acid, as well as electrochemical triggers can be utilized to shift the star polymers to the unbound state via CD/ferrocene complex dissociation. Moreover, heating above the lower critical solution temperature (approximately 34 °C) allows for a change from the coil to the globular state of the star polymers in the case of PDEA arms. For PDMA arms, heating to 70 °C allows shifting of the system to the unbound state. The response of the supramolecular star polymers is carefully studied via various methods including cyclic voltammetry (CV), DLS, and turbidimetry. Overall, the supramolecular star polymers can be transformed into predefined states via individually addressable temperature and/or redox stimuli, presenting a novel dual gated self-assembling supramolecular star polymer system in aqueous solution.
Co-reporter:Eva Blasco, Michael B. Sims, Anja S. Goldmann, Brent S. Sumerlin, and Christopher Barner-Kowollik
Macromolecules July 25, 2017 Volume 50(Issue 14) pp:5215-5215
Publication Date(Web):June 26, 2017
DOI:10.1021/acs.macromol.7b00465
The translation of small molecule chemistries into efficient methodologies for polymer functionalization spans several decades, enabling critical advances in soft matter materials synthesis with tailored and adaptive property profiles. The present Perspective explores—based on selected examples—50 years of innovation in polymer functionalization chemistries. These span a diverse set of chemistries based on activated esters, thiol–ene/yne processes, nucleophilic systems based on isocyanates, reactions driven by the formation of imines and oximes, ring-opening processes, cycloadditions, and—in a recent renaissance—multicomponent reactions. In addition, a wide variety of chain types and architectures have been modified based on the above chemistries, often with exquisite chemical control, highlighted by key examples. We conclude our journey through polymer functionalization with the—in our view—most critically required advances that have the potential to move from “science fiction” to “science fact”.
Co-reporter:Yoshi W. Marien;Paul H. M. Van Steenberge;Katrin B. Kockler;Marie-Françoise Reyniers;Guy B. Marin;Dagmar R. D'hooge
Polymer Chemistry (2010-Present) 2017 vol. 8(Issue 20) pp:3124-3128
Publication Date(Web):2017/05/23
DOI:10.1039/C7PY00412E
A fast method for the reliable estimation of the photodissociation quantum yield Φdiss is presented. Pulsed laser polymerization (PLP) experiments are performed at various pulse energies (1.5–6 mJ) and regression analysis is performed to the ratio of the peak heights identified in the size exclusion chromatography (SEC) trace. The high accuracy of the method is demonstrated for PLP initiated by 2,2-dimethoxy-2-phenylacetophenone (DMPA), considering in silico generated data including large theoretical errors (up to 20%). The method has also been successfully applied to experimental data of DMPA based isothermal PLP of n-butyl acrylate at 306 K, with an estimated Φdiss of 0.42 ± 0.04. In the long term, the method will facilitate the evaluation of current and the design of new highly efficient photoinitiators.
Co-reporter:
Macromolecular Theory and Simulations 2017 Volume 26(Issue 1) pp:
Publication Date(Web):2017/01/01
DOI:10.1002/mats.201600048
The importance of the development of kinetic modeling tools to mechanistically understand and design bulk and solution reversible addition fragmentation chain transfer (RAFT) polymerization is highlighted. Both deterministic and stochastic kinetic modeling methods are covered, considering a detailed reaction scheme and accounting for the impact of diffusional limitations on the reaction rates. A novel strategy is introduced to fundamentally calculate the diffusional contributions for the apparent RAFT addition and fragmentation rate coefficients. Next to literature examples, case studies are included to demonstrate that detailed theoretical studies are indispensable to completely map the effect of the polymerization conditions and RAFT agent reactivity on the control over microstructural properties and the overall polymerization time. Guidelines for future kinetic modeling activities are formulated to enhance joined theoretical and experimental research.
Co-reporter:Dr. Bernhard V. K. J. Schmidt; Dr. Christopher Barner-Kowollik
Angewandte Chemie 2017 Volume 129(Issue 29) pp:8417-8417
Publication Date(Web):2017/07/10
DOI:10.1002/ange.201703068
Cyclodextrin-Wirt-Gast-Chemie ist ein leistungsfähiger Design-Ansatz für dynamische und adaptive Materialien mit einstellbaren Eigenschaften. B. V. K. J. Schmidt und C. Barner-Kowollik schildern im Aufsatz auf S. 8468, wie komplexe supramolekulare Strukturen für Anwendungen von der Biomedizin bis zu selbstheilenden Materialien erzeugt werden können. Wie Wirt-Gast-Wechselwirkungen verschiedene molekulare Stränge verbinden, so vereinigt dieses Jubiläumsheft – anlässlich des 150-jährigen Bestehens der GDCh und des 100-jährigen Bestehens des RACI – Beispiele aus verschiedenen chemischen Teilgebieten von deutschen und australischen Autoren.
Co-reporter:Dr. Bernhard V. K. J. Schmidt; Dr. Christopher Barner-Kowollik
Angewandte Chemie 2017 Volume 129(Issue 29) pp:8468-8488
Publication Date(Web):2017/07/10
DOI:10.1002/ange.201612150
AbstractDynamische und adaptive Materialien sind leistungsfähige Konstrukte in der Polymerchemie mit einer Vielzahl von Anwendungen im Wirkstofftransport, in biologisch aktiven Systemen oder selbstheilenden Materialien. Sehr häufig werden dynamische Materialien mittels maßgeschneiderter Cyclodextrin(CD)-Wirt-Gast-Wechselwirkungen aufgebaut. Die präzise Einbindung dieser Wirt- und Gastbausteine ermöglicht die Bildung komplexer makromolekularer Strukturen, die für den Aufbau von Strukturen höherer Ordnung mit vordefinierten Funktionen verwendet werden können. So können dynamische Materialien mit außergewöhnlichen Eigenschaftsprofilen erzeugt werden. Hier wird die hierarchische Bildung komplexer Architekturen vom molekularen über den makromolekularen und den kolloidalen zum makroskopischen Bereich beschrieben. Dabei wird ein besonderes Augenmerk auf die Funktionalität und Responsivität der Strukturen auf äußere Felder insbesondere im biologischen Kontext gelegt.
Co-reporter: Dr. Christopher Barner-Kowollik
Angewandte Chemie 2017 Volume 129(Issue 29) pp:8420-8421
Publication Date(Web):2017/07/10
DOI:10.1002/ange.201704093
Sowohl Australien als auch Deutschland blicken auf eine lange Geschichte in der chemischen Forschung, doch Kooperationen zwischen den beiden Chemikergemeinschaften könnten noch erheblich intensiver werden. Dazu können gemeinsame Förderprogramme, die helfen, die Lücke zwischen Grundlagenforschung und industrieller Anwendung zu schließen, und themenspezifische Tagungen erheblich beitragen …” Lesen Sie mehr im Editorial von Christopher Barner-Kowollik.
Co-reporter:Markus M. Zieger;Patrick Mueller;Dr. Alexer S. Quick; Dr. Martin Wegener; Dr. Christopher Barner-Kowollik
Angewandte Chemie 2017 Volume 129(Issue 20) pp:5717-5721
Publication Date(Web):2017/05/08
DOI:10.1002/ange.201701593
AbstractBasierend auf einem neuartigen Photolack wurden 3D-Mikrostrukturen durch direktes Laserschreiben (DLW) hergestellt, die gezielt und vollständig spaltbar sind. Die Netzwerke bestehen ausschließlich aus reversiblen Bindungen, die unter Bestrahlung eines Phenacylsulfid-Vernetzers entstehen. In einer radikalfreien Stufenwachstumsreaktion zwischen den sich bildenden reaktiven Thioaldehyden und einer Thiol-Komponente bilden sich Disulfidbrücken. Die Reaktion wurde in Lösung durch ESI-MS nachgewiesen. Induziert wird die Spaltung durch einen Thiol-Disulfidbrücken-Austausch mit Dithiothreitol, der die geschriebenen Strukturen “löscht”. Die milde Spaltung des Disulfidbrücken-Netzwerkes ist z. B. orthogonal zu Acrylat-basierten DLW-Strukturen. Um diesen Aspekt hervorzuheben, wurden DLW-Strukturen hergestellt, die reversible Strukturelemente in nicht-reversible Acrylat-basierte Standardgerüste integrieren. Im Anschluss wurde die selektive Spaltung gezeigt. Die Erreichbarkeit hoher lateraler Auflösung wurde durch Anfertigen definierter Liniengitter mit Linienabständen bis 300 nm nachgewiesen.
Co-reporter: Dr. Christopher Barner-Kowollik
Angewandte Chemie International Edition 2017 Volume 56(Issue 29) pp:8304-8305
Publication Date(Web):2017/07/10
DOI:10.1002/anie.201704093
“… Both Australia and Germany have long traditions in the chemical sciences, however there is considerable scope to expand collaborations between the two chemical research communities. This can be achieved by collaborative funding opportunities, closing the gap between fundamental research and industrial applications, and targeted interactive symposia …” Read more in the Editorial by Christopher Barner-Kowollik.
Co-reporter:Dr. Bernhard V. K. J. Schmidt; Dr. Christopher Barner-Kowollik
Angewandte Chemie International Edition 2017 Volume 56(Issue 29) pp:8350-8369
Publication Date(Web):2017/07/10
DOI:10.1002/anie.201612150
AbstractDynamic and adaptive materials are powerful constructs in macromolecular and polymer chemistry with a wide array of applications in drug delivery, bioactive systems, and self-healing materials. Very often, dynamic materials are based on carefully tailored cyclodextrin host–guest interactions. The precise incorporation of these host and guest moieties into macromolecular building blocks allows the formation of complex macromolecular structures with predefined functions. Thus, dynamic materials with extraordinary adaptive property profiles—responsive to thermal, chemical, and photonic fields—become accessible. This Review explores the hierarchical formation of dynamic materials and complex macromolecular structures from the molecular via the macromolecular to the colloidal and macroscopic level, with a specific emphasis on the functionality and responsiveness of the assemblies, specifically in biological contexts.
Co-reporter:Markus M. Zieger;Patrick Mueller;Dr. Alexer S. Quick; Dr. Martin Wegener; Dr. Christopher Barner-Kowollik
Angewandte Chemie International Edition 2017 Volume 56(Issue 20) pp:5625-5629
Publication Date(Web):2017/05/08
DOI:10.1002/anie.201701593
AbstractUsing an advanced functional photoresist we introduce direct-laser-written (DLW) 3D microstructures capable of complete degradation on demand. The networks consist exclusively of reversible bonds, formed by irradiation of a phenacyl sulfide linker, giving disulfide bonds in a radical-free step-growth polymerization via a reactive thioaldehyde. The bond formation was verified in solution by ESI-MS. To induce cleavage, dithiothreitol causes a thiol–disulfide exchange, erasing the written structure. The mild cleavage of the disulfide network is highly orthogonal to other, for example, acrylate-based DLW structures. To emphasize this aspect, DLW structures were prepared incorporating reversible structural elements into a non-reversible acrylate-based standard scaffold, confirming subsequent selective cleavage. The high lateral resolution achievable was verified by the preparation of well-defined line gratings with line separations of down to 300 nm.
Co-reporter:Tanja K. Claus;Siham Telitel;Alexander Welle;Martin Bastmeyer;Andrew P. Vogt;Guillaume Delaittre
Chemical Communications 2017 vol. 53(Issue 10) pp:1599-1602
Publication Date(Web):2017/01/31
DOI:10.1039/C6CC09897E
We introduce a methodology to reversibly pattern planar surfaces via the light-induced dimerization of anthracenes, particularly involving a 9-triazolylanthracene motif. Specifically, we demonstrate that the visible light-induced forward reaction can be employed to pattern small molecule species as well as polymers in a spatially resolved fashion. Under UV irradiation, the generated patterns can be erased to regenerate reactive areas, which are then available for a new functionalization step. The dynamic change in surface chemistry is evidenced by ToF-SIMS.
Co-reporter:Yoshi W. Marien, Paul H. M. Van Steenberge, Christopher Barner-KowollikMarie-Françoise Reyniers, Guy B. Marin, Dagmar R. D’hooge
Macromolecules 2017 Volume 50(Issue 4) pp:
Publication Date(Web):February 13, 2017
DOI:10.1021/acs.macromol.6b02627
Complete pulsed laser polymerization (PLP) log-molar mass distributions (log-MMDs) are accurately simulated using kinetic Monte Carlo (kMC) modeling to gain not only knowledge on the propagation but also on the chain initiation and termination reactivity. The isothermal kMC model (306–325 K) accounts for diffusional limitations and all relevant elementary reactions, considering n-butyl acrylate, 2,2-dimethoxy-2-phenylacetophenone (DMPA), and a frequency of 500 s–1. The disparate reactivities toward vinylic bonds of the DMPA fragments are essential to ensure the well-defined multimodality of the log-MMDs necessary to identify consecutive inflection points. It is also illustrated that PLP log-MMD data can be used to test the validity of models for apparent termination rate coefficients at low monomer conversions and that kMC simulations are a powerful tool to track the chain growth of the different radical types between laser pulses and to identify dominant elementary reactions.
Co-reporter:Markus M. Zieger, Ognen Pop-Georgievski, Andres de los Santos Pereira, Elisseos Verveniotis, Corinna M. Preuss, Matthias Zorn, Bernd Reck, Anja S. Goldmann, Cesar Rodriguez-Emmenegger, and Christopher Barner-Kowollik
Langmuir 2017 Volume 33(Issue 3) pp:
Publication Date(Web):December 21, 2016
DOI:10.1021/acs.langmuir.6b03419
We introduce a newly designed catechol-based compound and its application for the preparation of homogeneous monomolecular layers as well as for robust assemblies on various substrates. The precisely defined cyclic catechol material (CyCat) was prepared from ortho-dimethoxybenzene in a phenolic resin-like synthesis and subsequent deprotection, featuring molecules with up to 32 catechol units. The CyCat’s chemical structure was carefully assessed via matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF), proton nuclear magnetic resonance (1H NMR), diffusion ordered spectroscopy (2D DOSY) and high resolution electrospray ionization mass spectrometry (ESI MS) experiments. The formation of colloidal aggregates of the CyCat material in alkaline solution was followed by dynamic light scattering (DLS) and further verified by dropcasting CyCat from solution on highly oriented pyrolytic graphite (HOPG), which was examined by Kelvin probe force microscopy (KPFM). The adsorption behavior of the CyCat to form monomolecular layers was investigated in real time by surface plasmon resonance (SPR). Formation of these thin CyCat layers (1.6–2.1 nm) on Au, SiO2 and TiO2 substrates was corroborated by spectroscopic ellipsometry (SE) and X-ray photoelectron spectroscopy (XPS). The prepared coating perfectly reflects the surface structure of the underlying substrate and does not exhibit CyCat colloidal aggregates as verified by atomic force microscopy (AFM). The functional nature of the prepared catechol monolayers was evidenced by reaction with 4-bromophenethylamine and bis(3-aminopropyl)-terminated poly(ethylene oxide) (PEO). Multilayer assemblies were prepared by a simple procedure of iterative immersion in solutions of CyCat and a multifunctional amine on Au, SiO2 and TiO2 substrates forming thicker coatings (up to 12 nm). Postmodification with small organic molecules was performed to covalently attach trifluoroacetyl, tetrazole and 2-bromo-2-methylpropanoyl moieties to the amine groups of the multilayer assembly coating. Furthermore, the versatility of the novel multilayer coating was underpinned by “grafting-to” of phenacyl sulfide–terminated PEO and “grafting-from” of poly(methyl methacrylate) via surface-initiated atom transfer radical polymerization (ATRP).
Co-reporter:Carolin Heiler, Janin T. Offenloch, Eva Blasco, and Christopher Barner-Kowollik
ACS Macro Letters 2017 Volume 6(Issue 1) pp:
Publication Date(Web):December 29, 2016
DOI:10.1021/acsmacrolett.6b00858
We pioneer the synthesis of fluorescent single chain nanoparticles (SCNPs) via UV-light induced folding based on tetrazole chemistry directly in pure water. Water-soluble photoreactive precursor polymers based on poly(acrylic acid) (PAA) bearing tetrazole, alkene and tetraethylene glycol monomethyl ether moieties, (PAAn(Tet/p-Mal/TEG)), or simply tetrazoles moieties, PAAn(Tet), were generated via RAFT polymerization. While tetrazole, ene, and acrylic acid containing polymers fold via dual nitrile imine-mediated tetrazole-ene cycloaddition (NITEC) as well as nitrile imine-carboxylic acid ligation (NICAL), tetrazole and acrylic acid only functional prepolymers fold exclusively via NICAL. A detailed study of the underpinning photochemistry of NITEC and NICAL is also included. The resulting water-soluble SCNPs were carefully characterized via analytical techniques such as NMR, UV–vis, and fluorescence spectroscopy, as well as SEC and DLS.
Co-reporter:Alexander M. Schenzel, Norbert Moszner, and Christopher Barner-Kowollik
ACS Macro Letters 2017 Volume 6(Issue 1) pp:
Publication Date(Web):December 14, 2016
DOI:10.1021/acsmacrolett.6b00934
We introduce a new concept for λ-orthogonal photocurable and degradable polymer networks based on disulfone cross-linkers. The methacrylate-based monomer mixture can be cured via irradiation with visible light (400–520 nm) due to a germanium-based initiator in 10 min. Subsequently, disassembly can be induced via the UV light (350–400 nm) triggered decomposition of a photogenerated amine (PGA) that cleaves the disulfone cross-links of the network completely via a substitution reaction. For the disulfone-based cross-linking, a new dimethacrylate monomer containing the disulfone moiety is synthesized. The cleavage of the S–S bond via a nucleophilic substitution is evidenced using 5 equiv of diethylamine as a nucleophile. In order to achieve an in situ degradation, a UV-degradable PGA is prepared, and its degradation upon UV irradiation as well as its stability under visible light are demonstrated. 1H NMR spectroscopy in solution revealed a complete degradation of the disulfone in the presence of 5 equiv of the PGA. Finally, a swollen network was prepared and successfully degraded upon UV irradiation within 4 h.
Co-reporter:Thomas Gegenhuber;Alexander M. Schenzel;Anja S. Goldmann;Per B. Zetterlund
Chemical Communications 2017 vol. 53(Issue 77) pp:10648-10651
Publication Date(Web):2017/09/26
DOI:10.1039/C7CC06347D
We introduce the facile synthesis of segmented copolymers by a catalyst-free Diels–Alder (DA) reaction at ambient temperature via step-growth and subsequent reversible addition fragmentation chain transfer (RAFT) polymerization. High molecular weight step-growth polymers are readily obtained (Mw = 40 000 g mol−1), featuring trithiocarbonate moieties in their chain, which allow monomer insertion via RAFT polymerization yielding high molecular weight species.
Co-reporter:Nicolai D. Knöfel;Hannah Rothfuss;Dr. Johannes Willenbacher; Dr. Christopher Barner-Kowollik; Dr. Peter W. Roesky
Angewandte Chemie 2017 Volume 129(Issue 18) pp:5213-5213
Publication Date(Web):2017/04/24
DOI:10.1002/ange.201702519
Wiederverwendbare platinhaltige Einzelketten-Nanopartikel (SCNPs), die ihre Form und Funktion bei Platin-Homogenkatalysen beibehalten, weisen den Weg zu neuartigen Katalysesystemen. In ihrer Zuschrift auf S. 5032 zeigen C. Barner-Kowollik, P. W. Roesky und Mitarbeiter anhand der Aminierung von Allylalkohol, dass die Platin(II)-SCNPs die höhere Aktivität von Homogenkatalysatoren mit der leichteren Zurückgewinnung von Heterogenkatalysatoren vereinen.
Co-reporter:Nicolai D. Knöfel;Hannah Rothfuss;Dr. Johannes Willenbacher; Dr. Christopher Barner-Kowollik; Dr. Peter W. Roesky
Angewandte Chemie 2017 Volume 129(Issue 18) pp:5032-5036
Publication Date(Web):2017/04/24
DOI:10.1002/ange.201700718
AbstractPlatin(II)-verknüpfte Einzelketten-Nanopartikel (PtII-SCNPs) wurden synthetisiert, charakterisiert und als wiederverwendbare Homogenkatalysatoren eingesetzt. Als Vorstufe wurde ein lineares Copolymer aus Styrol und 4-(Diphenylphosphan)styrol mittels Nitroxid-vermittelter Polymerisation synthetisiert. Die Triarylphosphaneinheiten, die sich im Polymerrückgrad befinden, ermöglichen durch Zugabe von [Pt(1,5-cyclooctadien)Cl2] in verdünnter Lösung die intramolekulare Vernetzung einzelner Ketten. Die Bildung von PtII-SCNPs wurde durch Gelpermeationschromatographie, dynamische Lichtstreuung, NMR-Spektroskopie (1H, 31P{1H}, 195Pt) und DOSY-Messungen nachgewiesen. Die erfolgreiche Verwendung der PtII-SCNPs als wiederverwendbare Homogenkatalysatoren wurde am Beispiel der Aminierung von Allylalkohol veranschaulicht.
Co-reporter:Dr. Bernhard V. K. J. Schmidt; Dr. Christopher Barner-Kowollik
Angewandte Chemie International Edition 2017 Volume 56(Issue 29) pp:8301-8301
Publication Date(Web):2017/07/10
DOI:10.1002/anie.201703068
Cyclodextrin-driven host–guest chemistry is a powerful approach for designing dynamic and adaptive materials with finely tunable properties. B. V. K. J. Schmidt and C. Barner-Kowollik describe in their Review on page 8350 ff. how complex supramolecular structures can be formed for applications ranging from biomedicine to self-healing materials. Akin to host–guest interactions that fuse different molecular strands, this Special Issue dedicated to 150 years of the GDCh and 100 years of the RACI fuses examples of German and Australian chemistry from a range of disciplines.
Co-reporter: Christopher Barner-Kowollik; Martin Bastmeyer;Dr. Eva Blasco;Dr. Guillaume Delaittre;Patrick Müller;Dr. Benjamin Richter; Martin Wegener
Angewandte Chemie 2017 Volume 129(Issue 50) pp:16038-16056
Publication Date(Web):2017/12/11
DOI:10.1002/ange.201704695
Abstract3D-Druck ist eine leistungsfähige Technik für die maßgeschneiderte Herstellung funktionaler Materialien. Dieser Aufsatz fasst den Stand der Technik im Hinblick auf 3D-Laser-Mikro- und -Nanodruck zusammen und erkundet die chemischen Herausforderungen, die derzeit die volle Etablierung dieser Technologie limitieren: von der Entwicklung fortgeschrittener Materialien für Anwendungen in der Zellbiologie und der Elektronik bis hin zu den bestehenden chemischen Grenzen des schnellen Schreibens mit Auflösungen unterhalb der Beugungsgrenze. Des Weiteren untersuchen wir Möglichkeiten zur Realisierung des direkten Laserschreibens mehrerer Materialien aus einem Photolack heraus, basierend auf wellenlängenselektiven photochemischen Prozessen (λ-Orthogonalität). Schließlich betrachten wir chemische Prozesse, mit deren Hilfe adaptive 3D-Strukturen geschrieben werden können, die auf externe Stimuli wie Licht, Wärme, pH-Wert oder spezifische Moleküle reagieren, sowie fortgeschrittene Konzepte für abbaubare Stützstrukturen.
Co-reporter:Eva Blasco;Jonathan Müller;Patrick Müller;Vanessa Trouillet;Markus Schön;Torsten Scherer;Martin Wegener
Advanced Materials 2016 Volume 28( Issue 18) pp:3592-3595
Publication Date(Web):
DOI:10.1002/adma.201506126
Co-reporter:Jan Steinkoenig, Hannah Rothfuss, Andrea Lauer, Bryan T. Tuten, and Christopher Barner-Kowollik
Journal of the American Chemical Society 2016 Volume 139(Issue 1) pp:51-54
Publication Date(Web):December 14, 2016
DOI:10.1021/jacs.6b10952
Herein, we introduce the first approach to map single-chain nanoparticle (SCNP) folding via high-resolution electrospray ionization mass spectrometry (ESI MS) coupled with size exclusion chromatography. For the first time, the successful collapse of polymeric chains into SCNPs is imaged by characteristic mass changes, providing detailed mechanistic information regarding the folding mechanism. As SCNP system we employed methyl methacrylate (MMA) statistically copolymerized with glycidyl methacrylate (GMA), resulting in p(MMA-stat-GMA), subsequently collapsed by using B(C6F5)3 as catalyst. Both the precursor polymer and the SCNPs can be well ionized via ESI MS, and the strong covalent cross-links are stable during ionization. Our high-resolution mass spectrometric approach can unambiguously differentiate between two mechanistic modes of chain collapse for every chain constituting the SCNP sample.
Co-reporter:Kai Hiltebrandt; Katharina Elies; Dagmar R. D’hooge; James P. Blinco
Journal of the American Chemical Society 2016 Volume 138(Issue 22) pp:7048-7054
Publication Date(Web):May 6, 2016
DOI:10.1021/jacs.6b01805
We introduce an efficient reaction manifold where the rate of a thermally induced ligation can be controlled by a photonic field via two competing reaction channels. The effectiveness of the reaction manifold is evidenced by following the transformations of macromolecular chain termini via high-resolution mass spectrometry and subsequently by selective block copolymer formation. The light-controlled reaction manifold consists of a so-called o-quinodimethane species, a photocaged diene, that reacts in the presence of light with suitable enes in a Diels–Alder reaction and undergoes a transformation into imines with amines in the absence of light. The chemical selectivity of the manifold is controlled by the amount of ene present in the reaction and can be adjusted from 100% imine formation (0% photo product) to 5% imine formation (95% photo product). The reported light-controlled reaction manifold is highly attractive because a simple external field is used to switch the selectivity of specific reaction channels.
Co-reporter:Martina M. Cecchini, Jan Steinkoenig, Samantha Reale, Leonie Barner, Jiayin Yuan, Anja S. Goldmann, Francesco De Angelis and Christopher Barner-Kowollik
Chemical Science 2016 vol. 7(Issue 8) pp:4912-4921
Publication Date(Web):21 Apr 2016
DOI:10.1039/C6SC01347C
We introduce a universal high resolution mass spectrometric method for the analysis of poly(ionic liquid)s (PILs), which belong to the most challenging polyelectrolytes from an analytical perspective, by fusing high resolution collision-induced dissociation (CID)-Orbitrap mass spectrometry (MS) with supercharging agents as well as quadrupole time-of-flight (QToF) MS. The study includes a wide array of hydrophilic halide-containing PILs, which were analyzed in negative mode. The influence of the core structures (based on imidazolium, triazolium, ammonium, phosphonium and pyridinium moieties), and variable styrene-, acrylate- and vinyl-type IL polymers on the ionization behavior is mapped in detail. Variable end group functionalities were introduced via functional chain transfer agents (CTA) in reversible addition-fragmentation chain transfer (RAFT) polymerization to study their behavior during the MS analysis. Furthermore, the demanding class of vinylimidazolium halide IL polymers was investigated. The current contribution thus introduces a new analytical technology platform for an entire polymer class.
Co-reporter:Antonina Vigovskaya, Doris Abt, Ishtiaq Ahmed, Christof M. Niemeyer, Christopher Barner-Kowollik and Ljiljana Fruk
Journal of Materials Chemistry A 2016 vol. 4(Issue 3) pp:442-449
Publication Date(Web):01 Dec 2015
DOI:10.1039/C5TB02207J
A photocaged diene is introduced at the 5′-end of oligonucleotides using the H-phosphonate approach. The photoenol-functionalized DNA is subsequently employed for the conjugation to a protein and the spatially controlled immobilization onto surfaces using a light-induced Diels–Alder cycloaddition. Fully functional protein–DNA conjugates and patterned DNA surfaces are obtained under mild irradiation conditions.
Co-reporter:Kai Hiltebrandt, Michael Kaupp, Edgar Molle, Jan P. Menzel, James P. Blinco and Christopher Barner-Kowollik
Chemical Communications 2016 vol. 52(Issue 60) pp:9426-9429
Publication Date(Web):28 Jun 2016
DOI:10.1039/C6CC03848D
We introduce a light induced sequence enabling λ-orthogonal star polymer formation via an arms-first approach, based on an α,ω-functional polymer carrying tetrazole and o-methyl benzaldehyde moieties, which upon irradiation can readily undergo cycloaddition with a trifunctional maleimide core. Depending on the wavelength, the telechelic strand can be attached to the core at either photo-reactive end.
Co-reporter:Nils Wedler-Jasinski, Nicolas Delbosc, Marie-Alice Virolleaud, Damien Montarnal, Alexander Welle, Leonie Barner, Andreas Walther, Julien Bernard and Christopher Barner-Kowollik
Chemical Communications 2016 vol. 52(Issue 56) pp:8753-8756
Publication Date(Web):20 Jun 2016
DOI:10.1039/C6CC03612K
We introduce recodable surfaces solely based on reversible artificial hydrogen bonding interactions. We show that a symmetrical oligoamide (SOA) attached to poly(methyl methacrylate) (PMMA) can be repeatedly immobilized and cleaved off spatially defined surface domains photochemically functionalized with asymmetric oligoamides (AOAs). The spatially resolved recodability is imaged and quantified via ToF-SIMS.
Co-reporter:Michael Kaupp, Kai Hiltebrandt, Vanessa Trouillet, Patrick Mueller, Alexander S. Quick, Martin Wegener and Christopher Barner-Kowollik
Chemical Communications 2016 vol. 52(Issue 9) pp:1975-1978
Publication Date(Web):10 Dec 2015
DOI:10.1039/C5CC09444E
A wavelength selective technique for light-induced network formation based on two photo-active moieties, namely ortho-methylbenzaldehyde and tetrazole is introduced. The network forming species are photo-reactive star polymers generated via reversible activation fragmentation chain transfer (RAFT) polymerization, allowing the network to be based on almost any vinylic monomer. Direct laser writing (DLW) allows to form any complex three-dimensional structure based on the photo-reactive star polymers.
Co-reporter:Hatice Mutlu and Christopher Barner-Kowollik
Polymer Chemistry 2016 vol. 7(Issue 12) pp:2272-2279
Publication Date(Web):22 Feb 2016
DOI:10.1039/C5PY01937K
A chain-shattering polymer system consisting of nontoxic, renewable resource-based monomers via acyclic diene metathesis (ADMET) chemistry is introduced. Amphiphilic triblock copolymers with apparent molecular weights in the range from 10 to 23 kDa are synthesized using a monofunctional polyethylene glycol monoacrylate, which acts as a selective chain-transfer agent during the polymerization process. Most importantly, the functional polymers possess repetitive midchain azobenzene moieties imparting them with self-immolative properties. By virtue of the enzyme degradable azobenzene chain elements, the amphiphilic macromolecules can be efficiently degraded via a self-immolative reaction into small fragments. The construction of the macromolecules along with their degradation is evidenced by nuclear magnetic resonance spectroscopy, electrospray ionization mass spectrometry and size exclusion chromatography. In addition, the triggered degradation leads to a strong reduction in the UV absorptivity of the polymeric material.
Co-reporter:Jan Steinkoenig, Fabian R. Bloesser, Birgit Huber, Alexander Welle, Vanessa Trouillet, Steffen M. Weidner, Leonie Barner, Peter W. Roesky, Jiayin Yuan, Anja S. Goldmann and Christopher Barner-Kowollik
Polymer Chemistry 2016 vol. 7(Issue 2) pp:451-461
Publication Date(Web):26 Oct 2015
DOI:10.1039/C5PY01320H
The preparation and characterization of poly(ionic liquid)s (PILs) bearing a polystyrene backbone via reversible addition fragmentation chain transfer (RAFT) polymerization and their photolithographic patterning on silicon wafers is reported. The controlled radical polymerization of the styrenic ionic liquid (IL) monomers ([BVBIM]X, X = Cl− or Tf2N−) by RAFT polymerization is investigated in detail. We provide a general synthetic tool to access this class of PILs with controlled molecular weight and relatively narrow molecular weight distribution (2000 g mol−1 ≤ Mn ≤ 10000 g mol−1 with dispersities between 1.4 and 1.3 for p([BVBIM]Cl); 2100 g mol−1 ≤ MP ≤ 14000 g mol−1 for p([BVBIM]Tf2N)). More importantly, we provide an in-depth characterization of the PILs and demonstrate a detailed mass spectrometric analysis via matrix-assisted laser desorption ionization (MALDI) as well as – for the first time for PILs – electrospray ionization mass spectrometry (ESI-MS). Importantly, p([BVBIM]Cl) and p([DMVBIM]Tf2N) were photochemically patterned on silicon wafers. Therefore, a RAFT agent carrying a photoactive group based on ortho-quinodimethane chemistry – more precisely photoenol chemistry – was photochemically linked for subsequent controlled radical polymerization of [BVBIM]Cl and [DMVBIM]Tf2N. The successful spatially-resolved photografting is evidenced by surface-sensitive characterization methods such as X-ray photoelectron spectroscopy (XPS) and time-of-flight secondary ion mass spectrometry (ToF-SIMS). The presented method allows for the functionalization of diverse surfaces with poly(ionic liquid)s.
Co-reporter:Kai Pahnke, Naomi L. Haworth, Josef Brandt, Uwe Paulmann, Christian Richter, Friedrich G. Schmidt, Albena Lederer, Michelle L. Coote and Christopher Barner-Kowollik
Polymer Chemistry 2016 vol. 7(Issue 19) pp:3244-3250
Publication Date(Web):14 Apr 2016
DOI:10.1039/C6PY00470A
We demonstrate a novel and ready to prepare thermoreversible hetero Diels–Alder dilinker on the basis of dithiooxalates, enabling the mild, rapid and catalyst-free linkage of diverse diene species under ambient conditions for applications in the fields of, for example, modular ligation, self-healing or recyclable materials and surface modification amongst others. The linker was studied using quantum chemical calculations, and experimentally in small molecular reactions via UV/Vis spectroscopy, mass spectrometry and NMR as well as in step-growth polymerizations with diene-difunctional building blocks – characterized via (temperature dependent) SEC and HT NMR – as an example for efficient polymer ligation.
Co-reporter:Katrin B. Kockler, Friederike Fleischhaker and Christopher Barner-Kowollik
Polymer Chemistry 2016 vol. 7(Issue 26) pp:4342-4351
Publication Date(Web):02 Jun 2016
DOI:10.1039/C6PY00867D
The Mark–Houwink–Kuhn–Sakurada parameters as well as Arrhenius parameters of the propagation rate coefficient for a new group of nitrogen-containing methacrylates were determined via triple detector SEC and pulsed laser polymerization–size exclusion chromatography, respectively. The data obtained for 2-(N-ethylanilino)ethyl methacrylate (NEAEMA, A = 1.77 (−0.75 to +5.74) × 106 L mol−1 s−1; EA = 20.17 (−2.57 to +2.87) kJ mol−1), 2-morpholinoethyl methacrylate (MOMA, A = 1.48 (−0.58 to +4.22) × 106 L mol−1 s−1; EA = 19.59 (−2.12 to +2.72) kJ mol−1), and 2-(1-piperidyl)ethyl methacrylate (PipEMA, A = 1.96 (−0.65 to +2.92) × 106 L mol−1 s−1; EA = 20.27 (−1.47 to +1.97) kJ mol−1) can be described with joint Arrhenius parameters of A = 1.83 (−0.72 to +3.65) × 106 L mol−1 s−1; EA = 20.14 (−2.17 to +2.28) kJ mol−1 and introduce a new family of nitrogen-containing branched methacrylates. The data of this novel family are critically evaluated and compared to the existing data sets for methacrylates with branched and cyclic ester side chains, respectively.
Co-reporter:Alexer M. Schenzel;Christopher Klein;Kai Rist;Norbert Moszner
Advanced Science 2016 Volume 3( Issue 3) pp:
Publication Date(Web):
DOI:10.1002/advs.201500361
Here, the development of an adhesive is reported – generated via free radical polymerization – which can be degraded upon thermal impact within minutes. The degradation is based on a stimuli responsive moiety (SRM) that is incorporated into the network. The selected SRM is a hetero Diels-Alder (HDA) moiety that features three key properties. First, the adhesive can be degraded at relatively low temperatures (≈80 °C), second the degradation occurs very rapidly (less than 3 min), and third, the degradation of the network can readily be analyzed and quantified due to its self-reporting nature. The new reversible self-reporting adhesion system is characterized in detail starting from molecular studies of the retro HDA reaction. Moreover, the mechanical properties of the network, as well as the adhesion forces, are investigated in detail and compared to common methacrylate-based systems, demonstrating a significant decrease in mechanic stability at elevated temperatures. The current study thus represents a significant advance of the current state of the art for debonding on demand adhesives, making the system interesting for several fields of application including dental adhesives.
Co-reporter:Christina M. Geiselhart, Janin T. Offenloch, Hatice Mutlu, and Christopher Barner-Kowollik
ACS Macro Letters 2016 Volume 5(Issue 10) pp:1146
Publication Date(Web):September 23, 2016
DOI:10.1021/acsmacrolett.6b00679
We introduce a facile and quantitative postpolymerization functionalization methodology for 1,4-polybutadienes, allowing us to decorate their pendent alkene functionalities with bromine and alkoxyether motifs carrying an array of functional groups ranging from tetrazoles to pyrenes. Specifically, the approach makes use of a mild, metal-free, electrophilic cascade reaction employing N-bromosuccinimide (NBS), a cyclic ether (i.e., THF), and a functional carboxylic acid. Detailed NMR, SEC, and ATR-IR studies confirm the successful modification.
Co-reporter:Josef Brandt, Naomi L. Haworth, Friedrich Georg Schmidt, Brigitte Voit, Michelle L. Coote, Christopher Barner-Kowollik, and Albena Lederer
ACS Macro Letters 2016 Volume 5(Issue 9) pp:1023
Publication Date(Web):August 15, 2016
DOI:10.1021/acsmacrolett.6b00551
We report an advanced analysis protocol that allows to quantitatively study the course of step-growth reactions by size exclusion chromatography on the example of the depolymerization of a Diels–Alder polymer based on a furane/maleimide couple at elevated temperatures. Frequently occurring issues of molar mass calibrations and overlap of monomer with solvent signals are addressed for determining reliable molar masses. Thereby, even kinetic parameters (e.g., rate coefficients) can be derived that otherwise would require performing additional spectroscopic experiments. Our results confirm first-order behavior of the rDA reaction with an activation energy of 33 kJ mol–1.
Co-reporter:Marcel Langer, Jan O. Mueller, Anja S. Goldmann, Felix H. Schacher, and Christopher Barner-Kowollik
ACS Macro Letters 2016 Volume 5(Issue 5) pp:597
Publication Date(Web):April 27, 2016
DOI:10.1021/acsmacrolett.6b00267
A dual functional chain transfer agent (CTA) capable of highly efficient sequential thermal and photoinduced ligation, generating α,ω-functional polymers, is introduced. The newly designed CTA (1-(4-((2-formyl-3-methyl phenoxy)methyl)phenyl)ethyl (diethoxyphosphoryl)methane dithioate) fuses thermally triggered hetero Diels–Alder chemistry with rapid light-induced photoenol chemistry. The versatility of the CTA is exemplarily demonstrated via the preparation of an amphiphilic triblock quaterpolymer poly(isoprene-co-styrene)-block-poly(ethyl acrylate)-block-poly(ethylene oxide) (P(I-co-S)-b-PEA-b-PEO). Subsequent to the homopolymerization of ethyl acrylate (PEA), a Cp-functional poly(isoprene-co-styrene) (P(I-co-S)) is conjugated with the electron-deficient C═S double bond (dienophile) of the CTA end group, generating a P(I-co-S)-b-PEA diblock terpolymer. The triblock quaterpolymer P(I-co-S)-b-PEA-b-PEO is generated by photoligation of a macromolecular dienophile, i.e., the fumarate-terminated poly(ethylene oxide) (PEO-fum) to the photoenol-functional P(I-co-S)-b-PEA. The new dual functional ligation RAFT agent constitutes a technology platform for generating α,ω-reactive building blocks from one single chain transfer agent.
Co-reporter:Thomas Tischer, Robert Gralla-Koser, Vanessa Trouillet, Leonie Barner, Christopher Barner-Kowollik, and Cornelia Lee-Thedieck
ACS Macro Letters 2016 Volume 5(Issue 4) pp:498
Publication Date(Web):March 30, 2016
DOI:10.1021/acsmacrolett.6b00106
Single molecule force spectroscopy (SMFS) is employed to gain insight into reversible addition–fragmentation chain transfer (RAFT) polymerization processes with living characteristics on glass surfaces. Surface-initiated (SI)-RAFT was selected to grow poly(hydroxyethyl methacrylate) (PHEMA). After aminolysis of the RAFT chain termini, thiol moieties serve as anchoring points for the gold tip of an atomic force microscope. The results allow to directly monitor the macromolecular growth of the surface-initiated polymerization. The obtained SMFS-based molecular weight distribution data of the polymers present on the surface indicate that the RAFT chain extension proceeds linearly with time up to high conversions. The current study thus adds SMFS as a valuable tool for the investigation of SI-RAFT polymerizations.
Co-reporter:Andrea Hufendiek, Anna Carlmark, Michael A. R. Meier, and Christopher Barner-Kowollik
ACS Macro Letters 2016 Volume 5(Issue 1) pp:139
Publication Date(Web):January 6, 2016
DOI:10.1021/acsmacrolett.5b00806
A facile light-induced procedure for the covalent cross-linking of cellulose at ambient conditions employing the nitrile imine mediated tetrazole-ene cycloaddition (NITEC) reaction is presented. Cellulose-tetrazoles with 2 degrees of substitution (0.14 and 0.23) were synthesized in a solution-based transesterification procedure in an ionic liquid. Two bismaleimides with either a trioxatridecane or a dithiodipropionyl backbone were used as cross-linkers to form fluorescent, covalently cross-linked cellulose networks and films, which were characterized by UV/vis spectroscopy, fluorescence spectroscopy, DSC, and TGA. The films showed a broad emission band from 500–700 nm and were thermally stable up to 200 °C. Using the bismaleimide with a disulfide moiety as the cross-linker, reductive degradation of the films can be induced. Finally, cellulose-tetrazole was cross-linked in a spatially resolved fashion, providing a strategy for the shaping of films based on renewable resources.
Co-reporter:Katrin B. Kockler;Alexer P. Haehnel;Thomas Junkers
Macromolecular Rapid Communications 2016 Volume 37( Issue 2) pp:123-134
Publication Date(Web):
DOI:10.1002/marc.201500503
Co-reporter:Ozcan Altintas
Macromolecular Rapid Communications 2016 Volume 37( Issue 1) pp:29-46
Publication Date(Web):
DOI:10.1002/marc.201500547
Co-reporter:Pieter Derboven;Paul H. M. Van Steenberge;Marie-Françoise Reyniers;Dagmar R. D'hooge;Guy B. Marin
Macromolecular Theory and Simulations 2016 Volume 25( Issue 2) pp:104-115
Publication Date(Web):
DOI:10.1002/mats.201500076
Co-reporter:Christopher Barner-Kowollik, Anja S. Goldmann, and Felix H. Schacher
Macromolecules 2016 Volume 49(Issue 14) pp:5001-5016
Publication Date(Web):June 29, 2016
DOI:10.1021/acs.macromol.6b00650
Polymer interfaces are ubiquitous in Nature and technology. Equipping artificial polymer-based interfaces with highly defined functions requires advanced macromolecular chemistry and powerful chemical tools. In this Perspective, we explore the nature of anisotropic—i.e., spatially resolved—polymer interfaces prepared via top-down and bottom-up approaches with selected examples from the recent literature. These range from self-assembly driven systems based on single polymer chains and block copolymers to lithographic encoding able to span wide spatial dimensions of patterning. Based thereon, we formulate the in our opinion required advances in polymer chemistry that will contribute significantly to preparing the next generation of structured interfaces. Among others, this includes the to-date limited orthogonality of parallelly executed ligation reactions as well as limits in λ-orthogonally addressable pericyclic ligation chemistry. Finally, we propose some long-term visions for not yet existing—however currently sought—technology that could drive interactive and adaptive polymer interface construction to new levels. These include the spatially resolved encoding of interfaces with molecular precision and the introduction of programmable properties to interfaces of varying shape and chemical complexity.
Co-reporter:Katrin B. Kockler, Friederike Fleischhaker, and Christopher Barner-Kowollik
Macromolecules 2016 Volume 49(Issue 22) pp:8572-8580
Publication Date(Web):November 11, 2016
DOI:10.1021/acs.macromol.6b01957
Nitrogen-containing methacrylates are a highly interesting class of monomers, yet only very limited data exist describing their propagation rate coefficients, kp. Herein, we investigate the propagation behavior of three N-containing monomers, namely 2-(N,N-diethylamino)ethyl methacrylate (DEAEMA), 2-(N,N-dimethylamino)ethyl methacrylate (DMAEMA), and 3-(N,N-dimethylamino)propyl methacrylate (DMAPMAE), where we systematically vary the ester side chain with respect to spacer and branching length with the aim of establishing if all so far investigated N-containing methacrylates display family type behavior with regard to kp. Thus, the Mark–Houwink–Kuhn–Sakurada parameters alongside the Arrhenius parameters of kp were determined for these monomers via triple detection SEC and pulsed laser polymerization–size exclusion chromatography (PLP-SEC). The obtained data result in Arrhenius parameters for DEAEMA of A = 2.07 (−0.79 to +3.98) × 106 L mol–1 s–1 and EA = 20.45 (−2.02 to +2.28) kJ mol–1, for DMAEMA of A = 2.64 (−0.79 to +1.98) × 106 L mol–1 s–1 and EA = 20.71 (−1.31 to +1.32) kJ mol–1, and for DMAPMAE of A = 1.22 (−0.54 to +8.02) × 106 L mol–1 s–1 and EA = 19.59 (−2.74 to +3.83) kJ mol–1. The data of the herein investigated monomers are critically compared to the previously published data of 2-(N-ethylanilino)ethyl methacrylate (NEAEMA), 2-(1-piperidyl)ethyl methacrylate (PipEMA), and 2-morpholinoethyl methacrylate (MOMA). It is found that DEAEMA and the previously investigated monomers can be described by one family, leading to a joint Arrhenius description for the four monomers NEAEMA, MOMA, PipEMA, and DEAEMA, best described by A = 1.55 (−0.57 to +3.88) × 106 L mol–1 s–1 and EA = 19.68 (−1.76 to +2.60) kJ mol–1.
Co-reporter:Elena Frick, Caroline Schweigert, Benjamin B. Noble, Hanna A. Ernst, Andrea Lauer, Yu Liang, Dominik Voll, Michelle L. Coote, Andreas-Neil Unterreiner, and Christopher Barner-Kowollik
Macromolecules 2016 Volume 49(Issue 1) pp:80-89
Publication Date(Web):November 13, 2015
DOI:10.1021/acs.macromol.5b02336
The fundamental influence of the structure and substitution of radical photoinitiators was investigated via a trifold combination of pulsed-laser polymerization with subsequent electrospray-ionization mass spectrometry (PLP-ESI-MS), femtosecond transient absorption (fs-TA) spectroscopy, and quantum chemistry. For the first time, a library of benzoin-derived photoinitiators with varied substitution patterns was synthesized. In the PLP-ESI-MS study, different photoinitiators were compared pairwise in so-called cocktail experiments, enabling the direct comparison of their initiation efficiency. In the fs-TA experiments, the transient response was observed after UV excitation in the visible spectral region, allowing for a description of excited state dynamics, which was analyzed with the aid of TD-DFT calculations. Ab initio calculations were undertaken to determine the reactivity of the radical fragments generated from these photoinitiators and to quantify the influence of various substituents on the rate of addition to monomer. In summary, the influence of the substituent on the initiation efficiency, intersystem crossing (ISC) behavior, excited state dynamics, and the extinction coefficients were analyzed. Hence, relaxation pathways and reaction mechanisms were optimized to explain disparate initiation efficiencies of a wide range of newly designed photoinitiators with varying substitution patterns. The strongly divergent absorptivities of the different photoinitiators and their corresponding initiation efficiencies underline that the absorptivity of a molecule is by no means an unequivocal measure for its reactivity when excited at a specific wavelength. In fact, the most efficient initiators are governed by one nπ* singlet state with a very low extinction coefficient at the excitation wavelength and one or two triplet states with nπ* character.
Co-reporter:Huey Wen Ooi, Benedikt Ketterer, Vanessa Trouillet, Matthias Franzreb, and Christopher Barner-Kowollik
Biomacromolecules 2016 Volume 17(Issue 1) pp:
Publication Date(Web):December 1, 2015
DOI:10.1021/acs.biomac.5b01391
We report the development of thermoresponsive 4-mercaptoethylpyridine (MEP)-based chromatographic microsphere based resins for antibody separation that show switchable release abilities by adsorbing immunoglobulins at 40 °C and releasing the proteins at 5 °C. The thermoswitchable release properties were introduced to the porous resins by the grafting of linear poly(N-isopropylacrylamide) (PNIPAM) chains synthesized via reversible addition–fragmentation chain transfer (RAFT) polymerization, which were modified to possess MEP end functionalities. Adsorption of γ-globulins as a model antibody on the shortest PNIPAM-MEP (3 kDa) grafted microparticles display binding capacities of up to 20 g L–1 at 40 °C and a significant decrease in binding capacity to less than 2.5 g L–1 at 5 °C. By switching the temperature to 5 °C, the release of bound γ-globulins is shown to be as high as 90%. The effects of polymer chain length on the binding capacity are studied in detail and found to be critical as they influence the density of MEP functionalities on the particle surfaces.
Co-reporter:Guillaume Delaittre, Nathalie K. Guimard, and Christopher Barner-Kowollik
Accounts of Chemical Research 2015 Volume 48(Issue 5) pp:1296
Publication Date(Web):April 14, 2015
DOI:10.1021/acs.accounts.5b00075
Synthetic polymer chemistry has undergone two major developments in the last two decades. About 20 years ago, reversible-deactivation radical polymerization processes started to give access to a wide range of polymeric architectures made from an almost infinite reservoir of functional building blocks. A few years later, the concept of click chemistry revolutionized the way polymer chemists approached synthetic routes. Among the few reactions that could qualify as click, the copper-catalyzed azide-alkyne cycloaddition (CuAAC) initially stood out. Soon, many old and new reactions, including cycloadditions, would further enrich the synthetic macromolecular chemistry toolbox. Whether click or not, cycloadditions are in any case powerful tools for designing polymeric materials in a modular fashion, with a high level of functionality and, sometimes, responsiveness.Here, we wish to describe cycloaddition methodologies that have been reported in the last 10 years in the context of macromolecular engineering, with a focus on those developed in our laboratories. The overarching structure of this Account is based on the three most commonly encountered cycloaddition subclasses in organic and macromolecular chemistry: 1,3-dipolar cycloadditions, (hetero-)Diels–Alder cycloadditions ((H)DAC), and [2+2] cycloadditions. Our goal is to briefly describe the relevant reaction conditions, the advantages and disadvantages, and the realized polymer applications. Furthermore, the orthogonality of most of these reactions is highlighted because it has proven highly beneficial for generating unique, multifunctional polymers in a one-pot reaction.The overview on 1,3-dipolar cycloadditions is mostly centered on the application of CuAAC as the most travelled route, by far. Besides illustrating the capacity of CuAAC to generate complex polymeric architectures, alternative 1,3-dipolar cycloadditions operating without the need for a catalyst are described. In the area of (H)DA cycloadditions, beyond the popular maleimide/furan couple, we present chemistries based on more reactive species, such as cyclopentadienyl or thiocarbonylthio moieties, particularly stressing the reversibility of these systems. In these two greater families, as well as in the last section on [2+2] cycloadditions, we highlight phototriggered chemistries as a powerful tool for spatially and temporally controlled materials synthesis.Clearly, cycloaddition chemistry already has and will continue to transform the field of polymer chemistry in the years to come. Applying this chemistry enables better control over polymer composition, the development of more complicated polymer architectures, the simplification of polymer library production, and the discovery of novel applications for all of these new polymers.
Co-reporter:Astrid F. Hirschbiel;Simone Geyer;Basit Yameen;Alexer Welle;Pavel Nikolov;Stefan Giselbrecht;Steffen Scholpp;Guillaume Delaittre
Advanced Materials 2015 Volume 27( Issue 16) pp:2621-2626
Publication Date(Web):
DOI:10.1002/adma.201500426
Co-reporter:Lutz Greb; Hatice Mutlu; Christopher Barner-Kowollik;Jean-Marie Lehn
Journal of the American Chemical Society 2015 Volume 138(Issue 4) pp:1142-1145
Publication Date(Web):December 23, 2015
DOI:10.1021/jacs.5b12198
N-alkyl α-bisimines were employed as main-chain functional groups in acyclic diene metathesis (ADMET)-polymers, conferring dual responsiveness for the controlled switching of the polymeric particle shape with light and metal ions. Photochemical Z/E-isomerization leads to a significant and reversible change in hydrodynamic volume, thus introducing simple imines as novel photoswitches for light-responsive materials. Mild imine-directed CH activation by Pd(OAc)2 is demonstrated as a new single-chain nanoparticle (SCNP) folding process, enabling a controlled atom- and step-economic SCNP synthesis. The combination of light- and metallo-responsiveness in the same polymer provides the ability for orthogonal switching, a valuable tool for advanced functional material design.
Co-reporter:Elena Frick; Athina Anastasaki; David M. Haddleton
Journal of the American Chemical Society 2015 Volume 137(Issue 21) pp:6889-6896
Publication Date(Web):May 13, 2015
DOI:10.1021/jacs.5b03048
The initiation mechanism of photochemically mediated Cu-based reversible-deactivation radical polymerization (photoRDRP) was investigated using pulsed-laser polymerization (PLP) and high-resolution mass spectrometry. The variation of the catalyst composition and ESI-MS analysis of the resulting products provided information on how initiator, ligand, copper species, and monomer are interacting upon irradiation with UV light. A discussion of the results allows for a new postulation of the mechanism of photoRDRP and–for the first time–the unambiguous identification of the initiating species and their interactions within the reaction mixture. One pathway for radical generation proceeds via UV light-induced C–Br bond scission of the initiator, giving rise to propagating radicals. The generation of copper(I) species from copper(II) can occur via several pathways, including, among others, via reduction by free amine ligand in its excited as well as from its ground state via the irradiation with UV light. The amine ligand serves as a strong reducing agent and is likely the main participant in the generation of copper(I) species.
Co-reporter:Alexer S. Quick;Andres de los Santos Pereira;Michael Bruns;Tiemo Bückmann;Cesar Rodriguez-Emmenegger;Martin Wegener
Advanced Functional Materials 2015 Volume 25( Issue 24) pp:3735-3744
Publication Date(Web):
DOI:10.1002/adfm.201500683
3D mesostructures with a height of up to 1 mm and micrometer feature size are fabricated employing a writing speed of 1 cm s−1 via direct laser writing utilizing a novel functional photoresist based on the radical coupling reaction of thiols and alkynes. The refractive index of the resist—consisting of a tetrafunctional thiol, a tetrafunctional alkyne and a photoinitiator—is tailored to be compatible with the employed high numerical aperture (NA) objective lens, thus enabling a Dip-in configuration. Mesostructures are characterized by scanning electron microscopy, optical photography, and nondestructive 3D time-of-flight secondary ion mass spectrometry. Woodpile photonic crystals are fabricated as benchmark structures in order to investigate the axial resolution. Verification of the chemical fabrication mechanism is achieved via transmission Fourier transform infrared (FTIR) spectroscopy of fabricated cuboid structures by monitoring the decrease of corresponding thiol and alkyne absorption peaks. Postmodification reactions, namely the thiol-Michael addition and the copper-catalyzed azide alkyne cycloaddition, are conducted employing residual thiols and alkynes throughout the cuboid structures. Successful dual and orthogonal modification throughout the structure and on the surface is achieved and verified utilizing transmission FTIR spectroscopy and time-of-flight secondary ion mass spectrometry.
Co-reporter:Kai Pahnke, Josef Brandt, Ganna Gryn'ova, Peter Lindner, Ralf Schweins, Friedrich Georg Schmidt, Albena Lederer, Michelle L. Coote and Christopher Barner-Kowollik
Chemical Science 2015 vol. 6(Issue 2) pp:1061-1074
Publication Date(Web):03 Nov 2014
DOI:10.1039/C4SC02908A
We report the investigation of fundamental entropic chain effects that enable the tuning of modular ligation chemistry – for example dynamic Diels–Alder (DA) reactions in materials applications – not only classically via the chemistry of the applied reaction sites, but also via the physical and steric properties of the molecules that are being joined. Having a substantial impact on the reaction equilibrium of the reversible ligation chemistry, these effects are important when transferring reactions from small molecule studies to larger or other entropically very dissimilar systems. The effects on the DA equilibrium and thus the temperature dependent degree of debonding (%debond) of different cyclopentadienyl (di-)functional poly(meth-)acrylate backbones (poly(methyl methacrylate), poly(iso-butyl methacrylate), poly(tert-butyl methacrylate), poly(iso-butyl acrylate), poly(n-butyl acrylate), poly(tert-butyl acrylate), poly(methyl acrylate) and poly(isobornyl acrylate)), linked via a difunctional cyanodithioester (CDTE) were examined via high temperature (HT) NMR spectroscopy as well as temperature dependent (TD) SEC measurements. A significant impact of not only chain mass and length with a difference in the degree of debonding of up to 30% for different lengths of macromonomers of the same polymer type but – remarkably – as well the chain stiffness with a difference in bonding degrees of nearly 20% for isomeric poly(butyl acrylates) is found. The results were predicted, reproduced and interpreted via quantum chemical calculations, leading to a better understanding of the underlying entropic principles.
Co-reporter:Lukas Stolzer, Alexander S. Quick, Doris Abt, Alexander Welle, Denys Naumenko, Marco Lazzarino, Martin Wegener, Christopher Barner-Kowollik and Ljiljana Fruk
Chemical Communications 2015 vol. 51(Issue 16) pp:3363-3366
Publication Date(Web):20 Jan 2015
DOI:10.1039/C4CC08880H
Photoreactive gold nanoparticles (NP) can be encoded in a spatially resolved fashion using direct laser writing techniques into variable patterns. The surface of the gold nanoparticles is imparted with photoreactivity by tethering photo-caged dienes (‘photoenols’), which are able to undergo a rapid Diels–Alder cycloaddition with surface anchored enes. Subsequent to surface encoding, the particles feature residual caged dienes, which can be reactivated for secondary surface encoding.
Co-reporter:Nicolas Zydziak, Florian Feist, Birgit Huber, Jan O. Mueller and Christopher Barner-Kowollik
Chemical Communications 2015 vol. 51(Issue 10) pp:1799-1802
Publication Date(Web):03 Dec 2014
DOI:10.1039/C4CC08756A
We report the first photochemical protocol for the generation of sequence defined macromolecules employing two hetero bifunctional photoreactive synthons, exploiting the orthogonal nature of photochemical – via the use of caged dienes – and thermally driven ligation protocols. We demonstrate that the iterative alternating synthon addition to an initial bifunctional core under irradiation at ambient temperature enables the generation of a macromolecule with up to 10 units (M = 3231.58 g mol−1, Đ = 1.00). The resulting macromolecules are monodisperse and feature absolute chain end fidelity. The unit-by-unit construction of the macromolecule is evidenced by Nuclear Magnetic Resonance Spectroscopy, Electrospray Ionization Mass Spectrometry and Size Exclusion Chromatography. The fundamental principle demonstrated herein paves the way for employing photochemical strategies for the design of sequence defined polymers.
Co-reporter:Yuuki Sugawara, Nils Jasinski, Michael Kaupp, Alexander Welle, Nicolas Zydziak, Eva Blasco and Christopher Barner-Kowollik
Chemical Communications 2015 vol. 51(Issue 65) pp:13000-13003
Publication Date(Web):09 Jul 2015
DOI:10.1039/C5CC05507E
An efficient methodology for modular fullerene functionalization via the photo-induced nitrile imine-mediated tetrazole–ene cycloaddition (NITEC) is introduced. The versatility and platform character of the method is illustrated by the light-driven reaction of fullerenes with small molecule, polymeric and surface-immobilized tetrazoles. The efficient fullerene conjugation is evidenced via mass spectrometric techniques.
Co-reporter:Astrid F. Hirschbiel, Waldemar Konrad, David Schulze-Sünninghausen, Steffen Wiedmann, Burkhard Luy, Bernhard V. K. J. Schmidt, and Christopher Barner-Kowollik
ACS Macro Letters 2015 Volume 4(Issue 10) pp:1062
Publication Date(Web):September 8, 2015
DOI:10.1021/acsmacrolett.5b00485
We combine supramolecular host–guest interactions of β-cyclodextrin (CD) with light-induced Diels–Alder reactions of 2-methoxy-6-methylbenzaldehyde (photoenol, PE) for the formation of multiblock copolymers. Via the synthesis of a new bifunctional chain transfer agent (CTA) and subsequent reversible addition–fragmentation chain transfer (RAFT) polymerization, we introduce a supramolecular recognition unit (tert-butyl phenyl) and a photoactive unit (photoenol) to a polymer chain in order to obtain an α,ω-functionalized polymeric center block, having orthogonal recognition units at each chain end. Multiblock copolymers are formed via the light-induced reaction of the photoenol with a maleimide-functionalized polymer chain and the supramolecular self-assembly of the tert-butyl phenyl group with the β-CD end group of a third polymer chain. By employing the fast and efficient photoinduced Diels–Alder reaction in combination with supramolecular host–guest interactions, a novel method for macromolecular modular ligation is introduced.
Co-reporter:Kai Pahnke, Ozcan Altintas, Friedrich G. Schmidt, and Christopher Barner-Kowollik
ACS Macro Letters 2015 Volume 4(Issue 7) pp:774
Publication Date(Web):July 7, 2015
DOI:10.1021/acsmacrolett.5b00335
We report the transfer of entropic chain length effects into the realm of supramolecular chemistry and thereby demonstrate a macromolecular method to tune the reaction equilibria of hydrogen bonding motifs via the application of substituents of differing lengths and masses while not altering the actual recognition units to achieve a difference in the degree of association. The supramolecular adducts are characterized via temperature-dependent nuclear magnetic resonance (NMR) spectroscopy.
Co-reporter:Ozcan Altintas, Thomas Josse, Julien De Winter, Nicholas M. Matsumoto, Pascal Gerbaux, Manfred Wilhelm and Christopher Barner-Kowollik
Polymer Chemistry 2015 vol. 6(Issue 39) pp:6931-6935
Publication Date(Web):24 Jul 2015
DOI:10.1039/C5PY01048A
We describe a simple and facile method for quantitatively converting bromine end-groups of well-defined polystyrene (PS, Mn,SEC = 4000 Da, Đ = 1.08) prepared by activators regenerated by electron transfer (ARGET) atom transfer radical polymerization (ATRP) into terminal alkenes by heating at 100 °C in dimethylformamide (DMF) without additional reagents. Subsequently, a facile quantitative post-functionalization of the terminal double bonds to various end functional polymers was performed via light-induced thiol–ene reactions. The quantitative end-group modifications as well as their thermal stability were assessed by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-ToF-MS), nuclear magnetic resonance (1H NMR) spectroscopy and size-exclusion chromatography (SEC), evidencing the generated functional polystyrenes to be highly stable up to 200 °C for extended periods of time (24 h).
Co-reporter:Johannes Willenbacher, Ozcan Altintas, Vanessa Trouillet, Nicolai Knöfel, Michael J. Monteiro, Peter W. Roesky and Christopher Barner-Kowollik
Polymer Chemistry 2015 vol. 6(Issue 24) pp:4358-4365
Publication Date(Web):23 Apr 2015
DOI:10.1039/C5PY00389J
We report the facile synthesis of well-defined palladium(II) cross-linked single-chain nanoparticles (Pd-SCNPs) using the ‘repeating unit approach’. The linear precursor polymer (Mn ≈ 10200 g mol−1, Đ ≈ 1.17) was synthesized via nitroxide mediated statistical copolymerization of styrene and 4-(chloromethyl)styrene (CMS) followed by a post-polymerization modification of the resulting copolymer to covalently attach the triarylphosphine ligand moieties. The ligand content along the lateral polymer chain was 12%. Intramolecular crosslinking was performed in diluted solution with a suitable precursor complex (Pd[1,5-cyclooctadiene]Cl2) to afford the well-defined Pd-SCNPs, which feature a hydrodynamic diameter of Dh = 5.4 nm. The palladium(II) containing single-chain nanoparticles were characterized in-depth using 1H NMR spectroscopy, 31P{1H} NMR spectroscopy, dynamic light scattering (DLS), size exclusion chromatography (SEC), 1H spin–spin relaxation time (T2) analysis, X-ray photoelectron spectroscopy (XPS), and log-normal distribution (LND) simulations. Finally, the applicability of the Pd-SCNPs as catalyst in the Sonogashira coupling was exemplified.
Co-reporter:Ozcan Altintas, Thomas Josse, Mahdi Abbasi, Julien De Winter, Vanessa Trouillet, Pascal Gerbaux, Manfred Wilhelm and Christopher Barner-Kowollik
Polymer Chemistry 2015 vol. 6(Issue 15) pp:2854-2868
Publication Date(Web):12 Feb 2015
DOI:10.1039/C5PY00036J
Linear polystyrenes carrying a mid-chain triazole, esters as well as terminal secondary bromines functionalities were synthesized via activators regenerated by electron transfer (ARGET) atom transfer radical polymerization (ATRP) using a bifunctional triazole containing initiator (3.8 kDa ≤ Mn,SEC ≤ 125 kDa, 1.08 ≤ Đ ≤ 1.19) with the aim of understanding their behavior under thermal and thermomechanical stress. As reference materials – isolating the influence of individual functional groups – three polystyrene homopolymers carrying an ω-bromine chain-end functionality, α,ω-ester-bromine functionalities as well as α,ω-dibromine/mid-chain ester functionalities (2 kDa ≤ Mn,SEC ≤ 39 kDa, 1.06 ≤ Đ ≤ 1.08) were prepared via ARGET ATRP. Furthermore, a well-defined triazole mid-chain functionalized block homopolymer, i.e. polystyrene-b-polystyrene (PS-b-PS, Mn,SEC = 4.4 kDa, Đ = 1.08), was synthesized via a combination of ARGET ATRP and copper(I)-catalyzed azide–alkyne cycloaddition (CuAAC) as a reference material. Reference polymers without bromine and with ester/triazole functionalities were additionally investigated. Thermomechanical stress was applied to the polymers via small scale extrusion as well as a rheological assessment (G′(t), G′′(t)) under processing conditions. The thermally challenged polymers were analyzed by size-exclusion chromatography (SEC), matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-ToF-MS), proton nuclear magnetic resonance (1H NMR) and X-ray photoelectron spectroscopy (XPS) to arrive at a detailed image of the degradation susceptibility of individual functional groups, especially esters, bromines and triazole functions. The findings indicate an enhanced degradation of ATRP polymers via an accelerated ester cleavage due to HBr release at high temperatures accompanied by a concomitant molecular weight increase due to the formation of triazolium salts via the reaction of triazole units with bromine terminal chain ends.
Co-reporter:Nils Jasinski, Andrea Lauer, Patrick J. M. Stals, Silke Behrens, Sarah Essig, Andreas Walther, Anja S. Goldmann, and Christopher Barner-Kowollik
ACS Macro Letters 2015 Volume 4(Issue 3) pp:298
Publication Date(Web):February 17, 2015
DOI:10.1021/acsmacrolett.5b00046
We introduce a novel electrochemical method for the purification of complex water-soluble functional polymers contaminated with copper salts originating from copper-catalyzed azide/alkyne ligation chemistry, for which no standard purification protocol is suitable. A triethylene glycol methyl ether methacrylate (TEGMA) star polymer with 2-ureido-4H-pyrimidone (UPy) end groups was prepared via an activator generated by electron transfer atom transfer radical polymerization (AGET ATRP) and copper-catalyzed azide/alkyne cycloaddition (CuAAc) and selected as a model system for electrolysis of an aqueous polymer solution. We systematically investigate the influence of sample concentration, voltage, and time of electrolysis on the quality of the purification. Atom emission spectroscopy (AES) reveals almost quantitative removal of copper, and size exclusion chromatography (SEC) as well as proton nuclear magnetic resonance spectroscopy (1H NMR) ensure the full integrity of the polymer under all selected conditions.
Co-reporter:Alexer P. Haehnel;Benjamin Wenn;Katrin Kockler;Tobias Bantle;Andrea M. Misske;Friederike Fleischhaker;Thomas Junkers
Macromolecular Rapid Communications 2015 Volume 36( Issue 22) pp:1984-1986
Publication Date(Web):
DOI:10.1002/marc.201500416
Co-reporter:Joachim Laun;Mariia Vorobii;Andres de los Santos Pereira;Ognen Pop-Georgievski;Vanessa Trouillet;Alexer Welle;Cesar Rodriguez-Emmenegger;Thomas Junkers
Macromolecular Rapid Communications 2015 Volume 36( Issue 18) pp:1681-1686
Publication Date(Web):
DOI:10.1002/marc.201500322
Co-reporter:Christoph Hörenz;Christian Pietsch;Anja S. Goldmann;Felix H. Schacher
Advanced Materials Interfaces 2015 Volume 2( Issue 8) pp:
Publication Date(Web):
DOI:10.1002/admi.201500042
Polymeric materials as building blocks represent the most important membrane materials as they are relatively simple to synthesize and flexible regarding manufacturing conditions. In this contribution, the synthesis of amphiphilic diblock terpolymers and their use for the preparation of integral asymmetric membranes via nonsolvent induced phase separation (NIPS) processes is presented. The diblock terpolymers consist of a hydrophobic poly(styrene-co-isoprene) block and a hydrophilic segment of poly(N,N-dimethylaminoethyl methacrylate). The materials are synthesized either via nitroxide mediated polymerization or living anionic polymerization. The NIPS process is used for the fabrication of porous diblock terpolymer membranes where the membrane morphology can be influenced by several parameters such as the applied solvent mixture, open time, or relative humidity. The resulting anisotropic membranes are characterized by scanning electron microscopy and water flux measurements. Furthermore, the UV-induced crosslinking of the isoprene part of the membrane matrix is demonstrated.
Co-reporter:Ozcan Altintas, David Schulze-Suenninghausen, Burkhard Luy, Christopher Barner-Kowollik
European Polymer Journal 2015 Volume 62() pp:409-417
Publication Date(Web):January 2015
DOI:10.1016/j.eurpolymj.2014.04.006
•First time design of miktoarm star polymers via H-bonding.•Extremely well defined building blocks.•Unambiguous characterization of the self-assembly.A supramolecular ABC-type miktoarm star polymer was prepared using a Hamilton wedge (HW) mid-chain functionalized polyethylene glycol-b-polystyrene (PEG-HW-PS) block copolymer and an α-cyanuric acid (CA) chain-end functional linear homopolymer poly-(n-butylacrylate) (CA-PnBA). The PEG-HW-PS element prepared via a combination of atom transfer radical polymerization (ATRP) and copper-catalyzed azide alkyne cycloaddition (CuAAC) (Mn = 13,700 g mol−1, PDI = 1.04). The CA-PnBA strand was synthesized via reversible addition–fragmentation chain transfer (RAFT) polymerization (Mn = 11,300 g mol−1, PDI = 1.06). The quantitative formation of a well-defined supramolecular ABC-type miktoarm star polymer was unambiguously proven via proton nuclear magnetic resonance (1H NMR) spectroscopy, diffusion ordered NMR spectroscopy (DOSY) and dynamic light scattering (DLS) analyses.
Co-reporter:Thomas Josse;Julien De Winter;Ozcan Altintas;Philippe Dubois;Pascal Gerbaux;Olivier Coulembier
Macromolecular Chemistry and Physics 2015 Volume 216( Issue 11) pp:1227-1234
Publication Date(Web):
DOI:10.1002/macp.201500054
Co-reporter:Katrin B. Kockler;Alexer P. Haehnel;Friederike Fleischhaker;Maria Schneider-Baumann;Andrea M. Misske
Macromolecular Chemistry and Physics 2015 Volume 216( Issue 14) pp:1573-1582
Publication Date(Web):
DOI:10.1002/macp.201500140
Co-reporter:Andrea Lauer, David E. Fast, Anne-Marie Kelterer, Elena Frick, Dmytro Neshchadin, Dominik Voll, Georg Gescheidt, and Christopher Barner-Kowollik
Macromolecules 2015 Volume 48(Issue 23) pp:8451-8460
Publication Date(Web):November 20, 2015
DOI:10.1021/acs.macromol.5b02127
The photostability of polymeric materials is crucial for their applicability, especially under potentially harsh environmental conditions. In the current study, the influence of methyl-substitution on the photochemical stability of photoinitiator-derived benzoyl end groups is systematically investigated by a combination of pulsed-laser polymerization and subsequent size exclusion chromatography coupled with electrospray ionization mass spectrometry (PLP–SEC–ESI–MS), chemically induced dynamic nuclear polarization–nuclear magnetic resonance spectroscopy (CIDNP–NMR), and density functional theory (DFT) calculations. Poly(methyl methacrylate)s (pMMA) were synthesized employing benzoin-type photoinitiators with systematically substituted benzoyl moieties (i.e., 2-methylbenzoin, 3-methylbenzoin, 4-methylbenzoin, 2,4-dimethylbenzoin, 2,6-dimethylbenzoin, 2,4,6-trimethylbenzoin, 2,3,5,6-tetramethylbenzoin, and 2,3,4,5,6-pentamethylbenzoin). Photoinduced cleavage of the photoinitiator-based end group (irradiation at 351 and 355 nm) occurs solely for polymeric species with benzoyl end groups carrying no or only one ortho-methyl substituent/s, whereas all of the other substitution patterns lead to stable chain termini. The theoretical calculations suggest that the different reactivity can be traced back to shifts of the n−π* transitions by approximately +0.25 eV. The current investigation unambiguously evidences that methylation in both ortho-positions of the benzoin-type photoinitiator critically enhances the photostability of the resulting polymer chain termini providing a clear instruction for photoinitiator design leading to polymers with stable chain termini.
Co-reporter:Ozcan Altintas, Müge Artar, Gijs ter Huurne, Ilja K. Voets, Anja R. A. Palmans, Christopher Barner-Kowollik, and E. W. Meijer
Macromolecules 2015 Volume 48(Issue 24) pp:8921-8932
Publication Date(Web):December 8, 2015
DOI:10.1021/acs.macromol.5b01990
We herein report the synthesis and characterization of ABC-type triblock copolymers containing two complementary association motifs and investigate their folding into well-defined polymeric nanoparticles under diluted conditions via intramolecular orthogonal hydrogen bonding. The precursor ABC-type triblock copolymers are prepared via reversible addition–fragmentation chain transfer (RAFT) polymerization bearing primary alkyl bromide on A, protected alkyne on B, and protected hydroxyl pendant groups on the C units. The dithioester groups of the RAFT polymers are quantitatively removed by radical-induced reduction before the side-chain functionalization. The complementary motifs, i.e., Hamilton wedge (HW, A block), benzene-1,3,5-tricarboxamide (BTA, B block), and cyanuric acid (CA, C block), are incorporated into the linear triblock copolymers side chains via postfunctionalization. The self-assembly processes of the HW and CA supramolecular motifs are followed by nuclear magnetic resonance (1H NMR) spectroscopy at ambient and elevated temperature in various solvents. The helical BTA stack formation is monitored by circular dichroism (CD) spectroscopy. In addition, the final aggregates formed by these two orthogonal forces, namely HW-CA pseudo-cross-linking and BTA stacking, are characterized by static and dynamic light scattering (SLS and DLS) as well as atomic force microscopy (AFM).
Co-reporter:Lukas Stolzer;Antonina Vigovskaya; Christopher Barner-Kowollik;Dr. Ljiljana Fruk
Chemistry - A European Journal 2015 Volume 21( Issue 41) pp:14309-14313
Publication Date(Web):
DOI:10.1002/chem.201502070
Abstract
A photochemical approach based on nitrile imine-mediated tetrazole-ene cycloaddition is introduced to functionalize gold nanorods with biomolecules. For this purpose, a bifunctional, photoreactive linker containing thioctic acid as the Au anchoring group and a tetrazole moiety for the light-induced reaction with maleimide-capped DNA was prepared. The tetrazole-based reaction on the nanoparticles’ surface results in a fluorescent pyrazoline product allowing for the spectroscopic monitoring of the reaction. This first example of nitrile imine-mediated tetrazole-ene cycloaddition (NITEC)-mediated biofunctionalization of Au nanorods paves the way for the attachment of sensitive biomolecules, such as antibodies and other proteins, under mild conditions and expands the toolbox for the tailoring of nanomaterials.
Co-reporter:Andres de los Santos Pereira, Nina Yu. Kostina, Michael Bruns, Cesar Rodriguez-Emmenegger, and Christopher Barner-Kowollik
Langmuir 2015 Volume 31(Issue 21) pp:5899-5907
Publication Date(Web):May 11, 2015
DOI:10.1021/acs.langmuir.5b01114
The precise design of bioactive surfaces, essential for the advancement of many biomedical applications, depends on achieving control of the surface architecture as well as on the ability to attach bioreceptors to antifouling surfaces. Herein, we report a facile avenue toward hierarchically structured antifouling polymer brushes of oligo(ethylene glycol) methacrylates via surface-initiated atom transfer radical polymerization (SI-ATRP) presenting photoactive tetrazole moieties, which permitted their functionalization via nitrile imine-mediated tetrazole-ene cyclocloaddition (NITEC). A maleimide-functional ATRP initiator was photoclicked to the side chains of a brush enabling a subsequent polymerization of carboxybetaine acrylamide to generate a micropatterned graft-on-graft polymer architecture as evidenced by X-ray photoelectron spectroscopy (XPS) and time-of-flight secondary ion mass spectrometry (ToF-SIMS). Furthermore, the spatially resolved biofunctionalization of the tetrazole-presenting brushes was accessed by the photoligation of biotin-maleimide and subsequent binding of streptavidin. The functionalized brushes bearing streptavidin were able to resist the fouling from blood plasma (90% reduction with respect to bare gold). Moreover, they were employed to demonstrate a model biosensor by immobilization of a biotinylated antibody and subsequent capture of an antigen as monitored in real time by surface plasmon resonance.
Co-reporter:Kai Hiltebrt;Dr. Thomas Pauloehrl;Dr. James P. Blinco;Katharina Linkert;Dr. Hans G. Börner;Dr. Christopher Barner-Kowollik
Angewandte Chemie International Edition 2015 Volume 54( Issue 9) pp:2838-2843
Publication Date(Web):
DOI:10.1002/anie.201410789
Abstract
A photochemical strategy enabling λ-orthogonal reactions is introduced to construct macromolecular architectures and to encode variable functional groups with site-selective precision into a single molecule by the choice of wavelength. λ-Orthogonal pericyclic reactions proceed independently of one another by the selection of functional groups that absorb light of specific wavelengths. The power of the new concept is shown by a one-pot reaction of equimolar quantities of maleimide with two polymers carrying different maleimide-reactive endgroups, that is, a photoactive diene (photoenol) and a nitrile imine (tetrazole). Under selective irradiation at λ=310–350 nm, any maleimide (or activated ene) end-capped compound reacts exclusively with the photoenol functional polymer. After complete conversion of the photoenol, subsequent irradiation at λ=270–310 nm activates the reaction of the tetrazole group with functional enes. The versatility of the approach is shown by λ-orthogonal click reactions of complex maleimides, functional enes, and polymers to the central polymer scaffold.
Co-reporter:Dr. Ozcan Altintas;Dr. Mathias Glassner;Dr. Cesar Rodriguez-Emmenegger;Dr. Alexer Welle;Vanessa Trouillet;Dr. Christopher Barner-Kowollik
Angewandte Chemie International Edition 2015 Volume 54( Issue 19) pp:5777-5783
Publication Date(Web):
DOI:10.1002/anie.201500485
Abstract
The efficient trapping of photogenerated thioaldehydes with functional shelf-stable nitrile oxides in a 1,3-dipolar cycloaddition is a novel and versatile photochemical strategy for polymer end-group functionalization and surface modification under mild and equimolar conditions. The modular ligation in solution was followed in detail by electrospray ionization mass spectrometry (ESI-MS). X-ray photoelectron spectroscopy (XPS) was employed to analyze the functionalized surfaces, whereas time-of-flight secondary-ion mass spectrometry (ToF-SIMS) confirmed the spatial control of the surface functionalization using a micropatterned shadow mask. Polymer brushes were grown from the surface in a spatially confined regime by surface-initiated atom transfer radical polymerization (SI-ATRP) as confirmed by TOF-SIMS, XPS as well as ellipsometry.
Co-reporter:Keita Fuchise, Peter Lindemann, Stefan Heißler, Hartmut Gliemann, Vanessa Trouillet, Alexander Welle, Jonathan Berson, Stefan Walheim, Thomas Schimmel, Michael A. R. Meier, and Christopher Barner-Kowollik
Langmuir 2015 Volume 31(Issue 10) pp:3242-3253
Publication Date(Web):February 23, 2015
DOI:10.1021/la505011j
The preparation of cross-linked nanosheets with 1–2 nm thickness and predefined shape was achieved by lithographic immobilization of trimethacryloyl thioalkanoates onto the surface of Si wafers, which were functionalized with 2-(phenacylthio)acetamido groups via a photoinduced reaction. Subsequent cross-linking via free radical polymerization as well as a phototriggered Diels–Alder reaction under mild conditions on the surface led to the desired nanosheets. Electrospray ionization mass spectrometry (ESI-MS), X-ray photoelectron spectroscopy (XPS), time-of-flight secondary ion mass spectrometry (ToF-SIMS), as well as infrared reflection-absorption spectroscopy (IRRAS) confirmed the success of individual surface-modification and cross-linking reactions. The thickness and lateral size of the cross-linked structures were determined by atomic force microscopy (AFM) for samples prepared on Si wafers functionalized with a self-assembled monolayer of 1H,1H,2H,2H-perfluorodecyl groups bearing circular pores obtained via a polymer blend lithographic approach, which led to the cross-linking reactions occurring in circular nanoareas (diameter of 50–640 nm) yielding an average thickness of 1.2 nm (radical cross-linking), 1.8 nm (radical cross-linking in the presence of 2,2,2-trifluoroethyl methacrylate as a comonomer), and 1.1 nm (photochemical cross-linking) of the nanosheets.
Co-reporter:Eva Blasco, Basit Yameen, Alexander S. Quick, Peter Krolla-Sidenstein, Alexander Welle, Martin Wegener, and Christopher Barner-Kowollik
Macromolecules 2015 Volume 48(Issue 24) pp:8718-8728
Publication Date(Web):December 4, 2015
DOI:10.1021/acs.macromol.5b02024
We report the synthesis and characterization of a new class of photoreactive conjugated polymers (Mn= 9300 g mol–1, Đ = 1.24; Mn= 9100 g mol–1, Đ = 1.23; Mn= 8800 g mol–1, Đ = 1.24) consisting of functionalized polythiophene-containing photoresponsive groups, i.e. a polythiophene containing 4-hydroxy-2,5-dimethylbenzophenone (DMBP) (“photoenol”) and/or maleimide groups. We evidence the chemical versatility and platform character of the generated light reactive polythiophenes in a range of examples by creating variable polythiophene functional 2D and 3D structures and morphologies. Single-chain nanoparticles and polymeric networks are formed by cross-linking of DMBP and maleimide containing polythiophene via UV light irradiation by varying the polymer concentration. Maleimide-functionalized polythiophene is employed for spatially resolved functionalization of surfaces and the coating of preformed 3D polymeric microstructures via direct laser writing (DLW). The generated polythiophene functional materials are carefully characterized via size exclusion chromatography, dynamic light scattering, and time-of-flight secondary ion mass spectrometry as well as FT-IR spectroscopy techniques.
Co-reporter:Kai Hiltebrt;Dr. Thomas Pauloehrl;Dr. James P. Blinco;Katharina Linkert;Dr. Hans G. Börner;Dr. Christopher Barner-Kowollik
Angewandte Chemie 2015 Volume 127( Issue 9) pp:2880-2885
Publication Date(Web):
DOI:10.1002/ange.201410789
Abstract
Eine photochemische Strategie nutzt λ-orthogonale Reaktionen zum Aufbau makromolekularer Architekturen und zum wellenlängenabhängigen Einbau chemischer Funktionalitäten in ein einzelnes Molekül. λ-Orthogonale pericyclische Reaktionen können unabhängig voneinander durch die spezifische photochemische Aktivierung einer chemischen Funktionalität ablaufen. Die Leistungsfähigkeit dieses neuen Konzeptes wird durch eine Eintopfreaktion belegt, bei der Maleinsäureimid mit zwei Polymeren, die unterschiedliche photoaktive Endgruppen (Photoenol und Tetrazol) tragen, selektiv reagieren. Beliebige aktivierte Doppelbindungen können durch eine gezielte Bestrahlung mit λ=310–350 nm mit einem Photoenol-funktionalisierten Polymer reagieren. Nach vollständigem Photoenolumsatz aktiviert eine Bestrahlung mit λ=270–310 nm die Reaktion des Tetrazols mit Maleimid. Die Vielseitigkeit dieses Ansatzes wird durch die λ-orthogonale Klick-Reaktionen von komplexen Maleimiden, funktionalen Enen und Polymeren an ein zentrales Polymergerüst gezeigt.
Co-reporter:Dr. Ozcan Altintas;Dr. Mathias Glassner;Dr. Cesar Rodriguez-Emmenegger;Dr. Alexer Welle;Vanessa Trouillet;Dr. Christopher Barner-Kowollik
Angewandte Chemie 2015 Volume 127( Issue 19) pp:5869-5875
Publication Date(Web):
DOI:10.1002/ange.201500485
Abstract
Das effiziente Abfangen photogenerierter Thioaldehyde durch funktionelle stabile Nitriloxide in einer 1,3-dipolaren Cycloaddition ist eine neue und vielseitige photochemische Strategie zur Funktionalisierung von Polymeren und zur Oberflächenmodifikation unter milden und äquimolaren Bedingungen. Die modulare Ligation in Lösung wird im Detail durch ESI-MS verfolgt. XPS wird zur Analyse der funktionalisierten Oberflächen verwendet, während ToF-SIMS die durch Anwendung einer Schattenmaske erreichte räumliche Kontrolle der Oberflächenfunktionalisierung belegt. Polymerbürsten wurden in einem räumlich begrenzten Bereich durch SI-ATRP von der Oberfläche aus wachsen gelassen, was mithilfe von ToF-SIMS, XPS sowie Ellipsometrie belegt wird.
Co-reporter:Astrid F. Hirschbiel, Bernhard V. K. J. Schmidt, Peter Krolla-Sidenstein, James P. Blinco, and Christopher Barner-Kowollik
Macromolecules 2015 Volume 48(Issue 13) pp:4410-4420
Publication Date(Web):June 24, 2015
DOI:10.1021/acs.macromol.5b00923
We introduce the design of a thermoresponsive nanoparticle via sacrificial micelle formation based on supramolecular host–guest chemistry. Reversible addition–fragmentation chain transfer (RAFT) polymerization was employed to synthesize well-defined polymer blocks of poly(N,N-dimethylacrylamide) (poly(DMAAm)) (Mn,SEC = 10 700 g mol–1, Đ = 1.3) and poly(N-isopropylacrylamide) (poly(NiPAAm)) (Mn,SEC = 39 700 g mol–1, Đ = 1.2), carrying supramolecular recognition units at the chain termini. Further, 2-methoxy-6-methylbenzaldehyde moieties (photoenols, PE) were statistically incorporated into the backbone of the poly(NiPAAm) block as photoactive cross-linking units. Host–guest interactions of adamantane (Ada) (at the terminus of the poly(NiPAAm/PE) chain) and β-cyclodextrin (CD) (attached to the poly(DMAAm chain end) result in a supramolecular diblock copolymer. In aqueous solution, the diblock copolymer undergoes micellization when heated above the lower critical solution temperature (LCST) of the thermoresponsive poly(NiPAAm/PE) chain, forming the core of the micelle. Via the addition of a 4-arm maleimide cross-linker and irradiation with UV light, the micelle is cross-linked in its core via the photoinduced Diels–Alder reaction of maleimide and PE units. The adamantyl–cyclodextrin linkage is subsequently cleaved by the destruction of the β-CD, affording narrowly distributed thermoresponsive nanoparticles with a trigger temperature close to 30 °C. Polymer chain analysis was performed via size exclusion chromatography (SEC), nuclear magnetic resonance (NMR) spectroscopy, and dynamic light scattering (DLS). The size and thermoresponsive behavior of the micelles and nanoparticles were investigated via DLS as well as atomic force microscopy (AFM).
Co-reporter:Pieter Derboven, Dagmar R. D’hooge, Marie-Francoise Reyniers, Guy B. Marin, and Christopher Barner-Kowollik
Macromolecules 2015 Volume 48(Issue 3) pp:492-501
Publication Date(Web):January 27, 2015
DOI:10.1021/ma5017659
Precision functionality is the key feature of next-generation radical polymerization enabling the implementation of applications such as controlled drug delivery, self-healing material design, and optoelectronic materials. The incorporation of functionality is however diametrically opposed to diffusion-controlled growth-inhibiting termination reactions. The fundamental bottleneck remains the identification of a generic and flexible protocol to accurately map the short–long termination reactivity. Herein, we introduce a unique framework based on the reversible addition–fragmentation chain transfer–chain length dependent–termination (RAFT-CLD-T) method that encompasses an extension of state-of-the-art fundamental theories and a correction for possible polymer matrix effects. Applied to methyl methacrylate (MMA) polymerization, the short–long termination reactivity is accurately quantified for the first time. Data analysis reveals the deficiency of currently used simplified models to describe the true short–long termination reactivity and the dominance of short-chain diffusivity. The proposed framework and insights are a turnkey prerequisite for the fundamental understanding of radical polymerization processes and to complete current macromolecular diffusion theories.
Co-reporter:Tao Wang;Dr. Yuzhou Wu;Dr. Seah Ling Kuan;Oliver Dumele;Dr. Markus Lamla;Dr. David Y. W. Ng;Matthias Arzt;Jessica Thomas;Jan O. Mueller ;Dr. Christopher Barner-Kowollik ;Dr. Tanja Weil
Chemistry - A European Journal 2015 Volume 21( Issue 1) pp:228-238
Publication Date(Web):
DOI:10.1002/chem.201403965
Abstract
A disulfide intercalator toolbox was developed for site-specific attachment of a broad variety of functional groups to proteins or peptides under mild, physiological conditions. The peptide hormone somatostatin (SST) served as model compound for intercalation into the available disulfide functionalization schemes starting from the intercalator or the reactive SST precursor before or after bioconjugation. A tetrazole–SST derivative was obtained that undergoes photoinduced cycloaddition in mammalian cells, which was monitored by live-cell imaging.
Co-reporter:Paul Lederhose, Naomi L. Haworth, Komba Thomas, Steven E. Bottle, Michelle L. Coote, Christopher Barner-Kowollik, and James P. Blinco
The Journal of Organic Chemistry 2015 Volume 80(Issue 16) pp:8009-8017
Publication Date(Web):July 13, 2015
DOI:10.1021/acs.joc.5b01088
The current study introduces a novel synthetic avenue for the preparation of profluorescent nitroxides via nitrile imine-mediated tetrazole-ene cycloaddition (NITEC). The photoinduced cycloaddition was performed under metal-free, mild conditions allowing the preparation of a library of the nitroxide functionalized pyrazolines and corresponding methoxyamines. High reaction rates and full conversion were observed, with the presence of the nitroxide having no significant impact on the cycloaddition performance. The formed products were investigated with respect to their photophysical properties in order to quantify their “switch on/off” behavior. The fluorescence quenching performance is strongly dependent on the distance between the chromophore and the free radical spin as demonstrated theoretically and experimentally. Highest levels of fluorescence quenching were achieved for pyrazolines with the nitroxide directly fused to the chromophore. Importantly, the pyrazoline profluorescent nitroxides were shown to efficiently act as sensors for redox/radical processes.
Co-reporter:Benjamin Vonhören, Marcel Langer, Doris Abt, Christopher Barner-Kowollik, and Bart Jan Ravoo
Langmuir 2015 Volume 31(Issue 50) pp:13625-13631
Publication Date(Web):November 24, 2015
DOI:10.1021/acs.langmuir.5b03924
Micropatterns of hydrophilic polymer brushes were prepared by micromolding in capillaries (MIMIC). The polymers are covalently bound to the surfaces by a rapid hetero Diels–Alder reaction, constituting the first example of polymers grafted to surfaces in a defined pattern by MIMIC. The polymers [poly(acrylic acid), poly(hydroxyethyl acrylate), and poly(tetraethylene glycol acrylate) ranging in molecular weight from 1500 to 6000 g mol–1] were prepared with narrow dispersities via the reversible addition–fragmentation chain transfer (RAFT) process using a highly electron deficient RAFT agent that can react with surface-anchored dienes such as cyclopentadiene. We demonstrate that the anchoring method is facile to perform and highly suitable for preparing patterned surfaces that are passivated against biological impact in well-defined areas.
Co-reporter:Bernhard V.K.J. Schmidt, Martin Hetzer, Helmut Ritter, Christopher Barner-Kowollik
Progress in Polymer Science 2014 Volume 39(Issue 1) pp:235-249
Publication Date(Web):January 2014
DOI:10.1016/j.progpolymsci.2013.09.006
The design of complex macromolecular architectures has driven macromolecular engineering over the past decades. The introduction of supramolecular chemistry into polymer chemistry provides novel opportunities for the generation of macromolecular architecture with specific functions. Cyclodextrins are attractive design elements as they form supramolecular inclusion complexes with hydrophobic guest molecules in aqueous solution affording the possibility to combine a large variety of building blocks to form novel macromolecular architectures. In the present critical review, the design of a broad range of macromolecular architectures driven by cyclodextrin host/guest chemistry is discussed, including supramolecular block copolymers, polymer brushes, star and branched polymers.
Co-reporter:Kim K. Oehlenschlaeger;Jan O. Mueller;Josef Brt;Stefan Hilf;Albena Lederer;Manfred Wilhelm;Robert Graf;Michelle L. Coote;Friedrich G. Schmidt
Advanced Materials 2014 Volume 26( Issue 21) pp:3561-3566
Publication Date(Web):
DOI:10.1002/adma.201306258
Co-reporter:Josef Brt;Kim K. Oehlenschlaeger;Friedrich Georg Schmidt;Albena Lederer
Advanced Materials 2014 Volume 26( Issue 33) pp:5758-5785
Publication Date(Web):
DOI:10.1002/adma.201400521
Dynamic bonding materials are of high interest in a variety of fields in material science. The reversible nature of certain reaction classes is frequently employed for introducing key material properties such as the capability to self-heal. In addition to the synthetic effort required for designing such materials, their analysis is a highly complex—yet important—endeavor. Herein, we critically review the current state of the art analytical methods and their application in the context of reversible bonding on demand soft matter material characterization for an in-depth performance assessment. The main analytical focus lies on the characterization at the molecular level.
Co-reporter:Thomas Tischer;Cesar Rodriguez-Emmenegger;Vanessa Trouillet;Alexer Welle;Vincent Schueler;Jan O. Mueller;Anja S. Goldmann;Eduard Brynda
Advanced Materials 2014 Volume 26( Issue 24) pp:4087-4092
Publication Date(Web):
DOI:10.1002/adma.201401006
Co-reporter:Michael Kaupp;Alexer S. Quick;Cesar Rodriguez-Emmenegger;Alexer Welle;Vanessa Trouillet;Ognen Pop-Georgievski;Martin Wegener
Advanced Functional Materials 2014 Volume 24( Issue 36) pp:5649-5661
Publication Date(Web):
DOI:10.1002/adfm.201400609
The synthesis and application of a novel reversible addition-fragmentation chain transfer (RAFT) agent carrying a photocaged thioaldehyde moiety is described (λmax = 355 nm). RAFT polymerization of styrene, dimethylacrylamide and a glycomonomer is evidenced (3600 g mol−1 ≤ Mn ≤ 15 000 g mol−1; 1.07 ≤ Đ ≤ 1.20) with excellent end-group fidelity. The photogenerated thioaldehyde on the chain ends can undergo hetero Diels–Alder reactions with dienes as well as reactions with nucleophiles. The terminal photoreactive polymers are photografted to porous diene-reactive polymeric microspheres. The grafted particles are in-depth characterized via scanning electron microscopy, elemental analysis, X-ray photoelectron spectroscopy, and high resolution FT-IR microscopy, leading to a qualitative as well as quantitative image of the core–shell objects. Grafting densities up to 0.10 molecules nm−2 are reached. The versatility of the thioaldehyde ligation is evidenced by spatially resolved grafting of polystyrene onto nucleophilic groups present in poly (dopamine) (PDA)-coated glass slides and silicon wafers via two-photon direct laser writing (DLW) imaged by ToF-SIMS. The combination of thioaldehyde ligation, RAFT polymerization, and DLW allows for the spatially resolved grafting of a vast range of polymers onto various substrates in any desired pattern with sub-micrometer resolution.
Co-reporter:Alexer S. Quick;Hannah Rothfuss;Alexer Welle;Benjamin Richter;Joachim Fischer;Martin Wegener
Advanced Functional Materials 2014 Volume 24( Issue 23) pp:3571-3580
Publication Date(Web):
DOI:10.1002/adfm.201304030
Three-dimensional microstructures are fabricated utilizing direct laser writing combined with a non-radical step polymerization based on multiphoton-induced Diels–Alder chemistry of o-quinodimethanes and maleimides. Woodpile photonic crystals with a total of five axial periods and a rod spacing of down to 500 nm are fabricated. The structures are characterized via scanning electron microscopy and focused ion beam milling. In addition, corresponding photonic stop bands are investigated via light microscopy as well as transmission and reflection spectroscopy. The Diels–Alder based network formation during direct laser writing is verified via infrared spectroscopy. Spatially resolved surface patterning of covalently bonded functional molecules on fabricated structures is demonstrated by employing the direct laser writing setup and a bromine containing maleimide. The successful surface modification is verified via time-of-flight secondary ion mass spectrometry.
Co-reporter:Daniel Volz, Astrid F. Hirschbiel, Daniel M. Zink, Jana Friedrichs, Martin Nieger, Thomas Baumann, Stefan Bräse and Christopher Barner-Kowollik
Journal of Materials Chemistry A 2014 vol. 2(Issue 8) pp:1457-1462
Publication Date(Web):04 Dec 2013
DOI:10.1039/C3TC32347A
The photoluminescence quantum efficiency as well as the processing properties of a series of brightly luminescent Cu(I)-metallopolymers strongly depended on the chosen synthetic approach. A monomeric, substituted styrenic complex features a photoluminescence quantum efficiency (PLQY) of only 4%, while its metallopolymeric thin film is over one order of magnitude more efficient.
Co-reporter:Andrew P. Vogt, Julien De Winter, Peter Krolla-Sidenstein, Udo Geckle, Olivier Coulembier and Christopher Barner-Kowollik
Journal of Materials Chemistry A 2014 vol. 2(Issue 23) pp:3578-3581
Publication Date(Web):04 Apr 2014
DOI:10.1039/C4TB00491D
A degradable polyphthalaldehyde-polystyrene block copolymer generated by modular ligation is reported for the first time serving as a nanochannel template for the formation of nanostructured materials. The polyphthalaldehyde-b-polystyrene copolymer was spin-coated onto a surface with subsequent polyphthalaldehyde block removal. Block conjugation and block removal were confirmed by H-NMR, SEC, AFM, and SEM.
Co-reporter:Corinna M. Preuss, Thomas Tischer, Cesar Rodriguez-Emmenegger, Markus M. Zieger, Michael Bruns, Anja S. Goldmann and Christopher Barner-Kowollik
Journal of Materials Chemistry A 2014 vol. 2(Issue 1) pp:36-40
Publication Date(Web):22 Oct 2013
DOI:10.1039/C3TB21317J
An avenue for the development of spatially resolved functional interfaces is presented. By introducing a novel, photo-reactive molecule – carrying a DOPA functionality and a photo-reactive group – we merge the ability of mussels to adhere to any surface with the spatial and temporal control of photo-click reactions, opening a plethora of applications in the biomedical and materials fields.
Co-reporter:Johannes Willenbacher, Bernhard V. K. J. Schmidt, David Schulze-Suenninghausen, Ozcan Altintas, Burkhard Luy, Guillaume Delaittre and Christopher Barner-Kowollik
Chemical Communications 2014 vol. 50(Issue 53) pp:7056-7059
Publication Date(Web):13 May 2014
DOI:10.1039/C4CC03218G
In the present communication we introduce a new platform technology for the reversible folding of single polymer chains in aqueous environment on the basis of cyclodextrin (CD) host–guest chemistry and controlled radical polymerization protocols. The single-chain folding of adamantyl-β-CD α-ω-functionalized poly(N,N-dimethylacrylamide) and its reversion at elevated temperatures were monitored by DLS and nuclear Overhauser enhancement spectroscopy (NOESY).
Co-reporter:Jan O. Mueller, Dominik Voll, Friedrich G. Schmidt, Guillaume Delaittre and Christopher Barner-Kowollik
Chemical Communications 2014 vol. 50(Issue 99) pp:15681-15684
Publication Date(Web):22 Oct 2014
DOI:10.1039/C4CC07792J
A facile, fast and ambient-temperature avenue towards highly fluorescent polymers is introduced via polymerizing non-fluorescent photoreactive monomers based on light-induced NITEC chemistry, providing a platform technology for fluorescent polymers. The resulting polypyrazolines were analyzed in depth and the photo-triggered step-growth process was monitored in a detailed kinetic study.
Co-reporter:Thomas Josse, Ozcan Altintas, Kim K. Oehlenschlaeger, Philippe Dubois, Pascal Gerbaux, Olivier Coulembier and Christopher Barner-Kowollik
Chemical Communications 2014 vol. 50(Issue 16) pp:2024-2026
Publication Date(Web):16 Dec 2013
DOI:10.1039/C3CC49067J
The light induced, catalyst-free ambient temperature preparation of macrocyclic aliphatic polyesters is pioneered. Based on the photo-induced Diels–Alder reaction of orthoquinodimethane and acrylate moieties, cyclic polyesters of high purity are readily synthesized. Considering the high tolerance to functional groups and the orthogonality of the ligation, the reported protocol can be easily transferred to a large range of polymers, complex topologies (tadpole, sun-shaped, jellyfish, etc.) and applications.
Co-reporter:Corinna M. Preuss, Markus M. Zieger, Cesar Rodriguez-Emmenegger, Nicolas Zydziak, Vanessa Trouillet, Anja S. Goldmann, and Christopher Barner-Kowollik
ACS Macro Letters 2014 Volume 3(Issue 11) pp:1169
Publication Date(Web):October 24, 2014
DOI:10.1021/mz5006469
We fuse the surface anchoring abilities of catechols with the rapid ligating nature of thiocarbonyl thio-based hetero-Diels–Alder (HDA) reactions via the synthesis of a new small molecule (HDA-DOPA-Cp) combining a HDA moiety with a catechol. Inspired by the mechanism of strong adhesion of marine mussels, we employed catechols as anchors to attach HDA ligation points to silicon wafers. The latter was exploited to generate a base for the HDA reactions on the surface employing α-cyclopentadiene (Cp) functional polymers such as poly(ethylene glycol)-Cp (PEG-Cp) and poly(trifluoro ethyl methacrylate)-Cp (PTFEMA-Cp) as dienes. By utilizing the fast and efficient HDA chemistry in combination with catechol anchoring groups, a new method for creating functional surfaces was developed.
Co-reporter:Johannes Willenbacher, Kilian N. R. Wuest, Jan O. Mueller, Michael Kaupp, Hans-Achim Wagenknecht, and Christopher Barner-Kowollik
ACS Macro Letters 2014 Volume 3(Issue 6) pp:574
Publication Date(Web):June 2, 2014
DOI:10.1021/mz500292e
We report the facile ambient temperature generation of size tunable and well-defined (pro)fluorescent single-chain nanoparticles (SCNPs) via the photoinduced nitrile imine intramolecular cross-ligation of linear precursor polymers, constituting a platform technology as novel imaging agents. A set of three linear precursor polymers (Mn ≈ 14000 g mol–1, Đ ≈ 1.25) was synthesized via nitroxide-mediated statistical copolymerization of styrene and 4-(chloromethyl)styrene (CMS), followed by a postpolymerization modification of the resulting copolymer installing protected maleimide (PG-Mal) as well as tetrazole (Tet) moieties. The tetrazole content (% Tet) along the lateral polymer chains was varied between 12 and 24% in order to preselect not only the size of the corresponding SCNPs, but also their fluorescence and reactive properties. Finally, the applicability of the profluorescent SCNPs for fluorescence labeling was demonstrated utilizing residual surface expressed Tet moieties on the SCNPs surface in a reaction with maleimide functional polymeric microspheres. The (pro)fluorescent single-chain nanoparticles were in-depth characterized by 1H NMR spectroscopy, dynamic light scattering (DLS), size exclusion chromatography (SEC), and atomic force microscopy (AFM), as well as UV/vis and fluorescence spectroscopy.
Co-reporter:Peter Gerstel, Stefanie Klumpp, Frank Hennrich, Angela Poschlad, Velimir Meded, Eva Blasco, Wolfgang Wenzel, Manfred M. Kappes, and Christopher Barner-Kowollik
ACS Macro Letters 2014 Volume 3(Issue 1) pp:10
Publication Date(Web):December 11, 2013
DOI:10.1021/mz400472q
Fourteen different “hairy-rod” conjugated polymers, 9,9-dioctylfluorene derivatives entailing 1,2,3-triazole, azomethine, ethynyle, biphenyle, stilbene, and azobenzene lateral units, are synthesized via modular conjugation and are systematically investigated with respect to their ability to selectively disperse SWCNTs. Four polymers of the azomethine type, with unprecedented selectivity toward dispersing (8,7), (7,6), and (9,5) SWCNT species, have been identified. In particular, azomethine polymers, herein applied for the first time for SWCNT dispersion, have been evidenced to be very effective in the highly selective solubilization of SWCNTs. The experimentally observed selectivity results are unambiguously supported by molecular dynamics simulations that account for the geometrical properties and deformation energy landscape of the polymer. Specifically, the calculations accurately and with high precision predict the experimentally observed selectivity for the (7,6) and (9,5) conformations.
Co-reporter:Marcel Langer, Josef Brandt, Albena Lederer, Anja S. Goldmann, Felix H. Schacher and Christopher Barner-Kowollik
Polymer Chemistry 2014 vol. 5(Issue 18) pp:5330-5338
Publication Date(Web):03 Jun 2014
DOI:10.1039/C4PY00644E
The present article reports the preparation of a novel class of switchable amphiphilic diblock copolymers with a temperature switchable linkage. Reversible addition fragmentation chain transfer (RAFT) polymerization was used to synthesize the individual blocks: for the preparation of the non-polar block, i.e. poly(isoprene-co-styrene) (P(I-co-S)) (9200 g mol−1 ≤ Mn ≤ 50000 g mol−1, 1.22 ≤ Đ ≤ 1.36), a chain transfer agent (CTA, 3-((2-bromo-2-methylpropanoyl)oxy)propyl 2-(((dodecylthio)carbonothioyl)thio)-2-methylpropanoate) carrying a bromine group was employed, ready for subsequent cyclopentadienyl (Cp) transformation. For the preparation of the polar block, triethylene glycol methyl ether acrylate (TEGA) was polymerized (6600 g mol−1 ≤ Mn ≤ 35000 g mol−1, 1.12 ≤ Đ ≤ 1.30) using a RAFT agent carrying a phosphoryl Z-group, which is able to undergo hetero Diels-Alder (HDA) ligation with Cp moieties. Both building blocks were conjugated at ambient temperature in the presence of ZnCl2 as catalyst yielding the amphiphilic block copolymer P(I-co-S)-b-PTEGA (16000 g mol−1 ≤ Mn ≤ 68000 g mol−1, 1.15 ≤ Đ ≤ 1.32). To investigate the bonding/debonding capability of the HDA linkage, high temperature nuclear magnetic resonance (HT-NMR) spectroscopy, high temperature dynamic light scattering (HT-DLS) and high temperature size exclusion chromatography (HT-SEC) were carried out, evidencing that efficiently switchable amphiphilic block copolymers were generated (>4 cycles).
Co-reporter:Ozcan Altintas, Mahdi Abbasi, Kamran Riazi, Anja S. Goldmann, Nico Dingenouts, Manfred Wilhelm and Christopher Barner-Kowollik
Polymer Chemistry 2014 vol. 5(Issue 17) pp:5009-5019
Publication Date(Web):08 May 2014
DOI:10.1039/C4PY00484A
Well-defined three-arm and four-arm star polymers designed via a Z-group approach carrying trithiocarbonate functionalities at the core are prepared via reversible addition-fragmentation chain transfer (RAFT) polymerization featuring molecular weights of Mn,SEC = 156 kDa, Đ = 1.16 (3-arm) and Mn,SEC = 162 kDa, Đ = 1.15 (4-arm) based on multi-angle laser light scattering (MALLS) detection, respectively. The star-shaped polystyrenes are subjected (in bulk) to thermal stress in the temperature range between 140 and 200 °C from 10 minutes up to 96 h. The thermally treated 3-arm and 4-arm star polymers are analyzed via size exclusion chromatography (SEC) to quantify the degradation process at variable temperatures as a function of time under an argon atmosphere. Cleavage rate coefficients of the star polymers are deduced as a function of temperature, resulting in activation parameters for the cleavage process, i.e. Ea = 131 kJ mol−1; A = 3.93 × 1011 s−1 (Mn,SEC = 156 kDa, Đ = 1.16, 3-arm star) and Ea, = 134 kJ mol−1; A = 9.13 × 1011 s−1 (Mn,SEC = 162 kDa, Đ = 1.15, 4-arm star), respectively. Processing of the star-shaped polymers is mimicked via a small scale counter rotating twin screw extrusion to achieve nonlinear shear and elongation flow under pressure. Furthermore, a rheological assessment via the linear shear deformation region (small amplitude oscillatory shear, SAOS) allows for a correlation of the processing conditions with the thermal degradation properties of the star polymers in the melt. Zero shear viscosity (η0) as a criterion of the degradation process is measured in the rheometer and correlated to the weight-average molecular weight, Mw.
Co-reporter:Jan O. Mueller, Nathalie K. Guimard, Kim K. Oehlenschlaeger, Friedrich G. Schmidt and Christopher Barner-Kowollik
Polymer Chemistry 2014 vol. 5(Issue 4) pp:1447-1456
Publication Date(Web):31 Oct 2013
DOI:10.1039/C3PY01381B
The efficient sunlight-induced crosslinking of 1,2-polybutadienes to generate fluorescent patterns with spatial resolution is reported. The photochemical conjugation method employed is based on a nitrile imine-mediated tetrazole–ene cycloaddition (NITEC) reaction, which proceeds under UV-light irradiation (λmax = 312 nm) at ambient temperature in the absence of any catalyst. The NITEC reaction between 1-pentene and a newly designed di-linker, consisting of two photosensitive diaryl-substituted tetrazoles joined by a tetraethylene glycol spacer, was investigated in an initial study. Detailed characterization of a small molecule model study was performed by size exclusion chromatography (SEC), UV-vis and fluorescence spectroscopy as well as electrospray-ionization mass spectrometry (ESI-MS), which was also employed for monitoring the progress of the reaction (100% conversion in 20 min). Finally, two 1,2-polybutadienes of disparate molar masses were each photocrosslinked with the di-linker. The crosslinking reaction parameters, such as concentration, di-linker fraction and reaction time were optimized via SEC analysis and gravimetric determination of gel fractions. The applicability of the novel crosslinking technology for generating spatially controlled highly fluorescent gel patterns is demonstrated in a solvent-free reaction for 2 h under sunlight. In summary, the current study introduces an efficient light-triggered technology platform for crosslinking polymers carrying non-activated double bonds.
Co-reporter:Alexander P. Haehnel, Marek Stach, Anna Chovancová, Jannick M. Rueb, Guillaume Delaittre, Andrea M. Misske, Igor Lacík and Christopher Barner-Kowollik
Polymer Chemistry 2014 vol. 5(Issue 3) pp:862-873
Publication Date(Web):03 Sep 2013
DOI:10.1039/C3PY00948C
The Arrhenius parameters of the propagation rate coefficient for two hetero-atom containing (meth)-acrylates (studied as 1 M solution in N,N-dimethylacetamide (DMAc)) are determined via the pulsed laser polymerization – size-exclusion chromatography (PLP-SEC) method. Absolute molar mass determination is achieved via SEC coupled to on-line multi-angle laser light scattering (MALLS). The data obtained for hydroxypropylcarbamate acrylate (HPCA, A = 3.97 (−1.44 to 1.63) × 106 L mol−1 s−1 and Ea = 14.3 (−1.38 to 5.13) kJ mol−1) are critically compared with the literature known data sets of two structural derivatives, i.e., 2-(phenylcarbamoyloxy)isopropyl acrylate (PhCPA) and 2-(hexylcarbamoyloxy)isopropyl acrylate (HCPA), indicating an increase in the propagation rate coefficient with increasing ester side chain length. Ureidoethyl methacrylate (UMA, A = 2.08 (−0.45 to 0.91) × 106 L mol−1 s−1 and Ea = 19.9 (−0.89 to 0.91) kJ mol−1) represents the first hetero-atom containing methacrylate to be studied via PLP-SEC, evidencing a significantly higher propagation rate coefficient compared to earlier investigated methacrylate-type monomers. Furthermore, the free-radical polymerization behavior of HPCA and UMA is studied via in situ1H-NMR experiments at elevated temperatures allowing for an estimation of average termination rate coefficients (at low conversion) in conjunction with the determined kp data. Furthermore, the polymerization of UMA was successfully controlled by reversible addition–fragmentation chain transfer (RAFT) polymerization as evidenced by the linear evolution of the number-average molar mass, Mn, with conversion (3000 g mol−1 ≤ Mn ≤ 23000 g mol−1, 1.15 ≤ Đ ≤ 1.3) as well as by nitroxide-mediated polymerization (NMP), as demonstrated by the linear evolution of Mn with conversion (4000 g mol−1 ≤ Mn ≤ 40000 g mol−1, 1.3 ≤ Đ ≤ 1.4). In addition, HPCA polymerization was successfully controlled by the RAFT process, as evidenced by the linear evolution of Mn with conversion (2000 g mol−1 ≤ Mn ≤ 21000 g mol−1, 1.2 ≤ Đ ≤ 1.4) and successful chain extension experiments. Finally, the NMP of HPCA exhibited uniform shifts of the molar mass distributions in the range of 5000 g mol−1 ≤ Mn ≤ 70000 g mol−1 and successful chain extension experiments.
Co-reporter:Bernhard V. K. J. Schmidt and Christopher Barner-Kowollik
Polymer Chemistry 2014 vol. 5(Issue 7) pp:2461-2472
Publication Date(Web):19 Dec 2013
DOI:10.1039/C3PY01580G
A wide variety of novel complex macromolecular X- and H-shaped star block copolymers based on supramolecular cyclodextrin (CD)/adamantane interactions are reported. Mid-chain adamantyl poly(N,N-dimethylacrylamide) (PDMA; Mn = 12000 g mol−1, Đ = 1.10), poly(N,N-diethylacrylamide) (PDEA; Mn = 5400 g mol−1, Đ = 1.08) and P(DEA-b-DMA) (Mn = 10400 g mol−1, Đ = 1.13) as well as double adamantyl end functionalized PDMA (Mn = 6400 g mol−1, Đ = 1.06) and PDEA (Mn = 6800 g mol−1, Đ = 1.11) are synthesized via reversible addition–fragmentation chain transfer (RAFT) polymerization. Furthermore, mid-chain β-CD functionalized PDMA (Mn = 10200 g mol−1, Đ = 1.26), PDEA (Mn = 11100 g mol−1, Đ = 1.30) and P(DEA-b-DMA) (Mn = 7600 g mol−1, Đ = 1.26) are synthesized via a combination of RAFT polymerization and copper(I) catalyzed azide alkyne cycloaddition. Subsequently, the formation of the target architectures via supramolecular association of CD- and adamantyl-functionalized building blocks is evidenced via nuclear Overhauser enhancement spectroscopy and dynamic light scattering. Furthermore, the temperature responsive behavior of the formed star block copolymers is investigated.
Co-reporter:Elena Frick, Hanna A. Ernst, Dominik Voll, Thomas J. A. Wolf, Andreas-Neil Unterreiner and Christopher Barner-Kowollik
Polymer Chemistry 2014 vol. 5(Issue 17) pp:5053-5068
Publication Date(Web):29 Apr 2014
DOI:10.1039/C4PY00418C
The initiation efficiency and the excited state dynamics of the three triplet radical photoinitiators MMMP (2-methyl-4′-(methylthio)-2-morpholinopropiophenone), benzoin (2-hydroxy-1,2-diphenylethanone, Bz), and 4-methyl benzoin (2-hydroxy-2-phenyl-1-(p-tolyl) ethanone, 4MB) are investigated via a trifold combination of pulsed laser polymerization and subsequent electrospray ionization mass spectrometry (PLP-ESI-MS), femtosecond transient absorption (TA) spectroscopy, and density functional theory (DFT) methods. A quantitative elucidation of the underlying mechanisms that cause unequal initiation efficiencies of the three photoinitiators is proposed suggesting higher radical formation capabilities for MMMP and 4MB compared to Bz based on the inherent photophysical properties of the three initiators. MMMP shows significantly higher complexity of the relaxation pathways due to partial excitation into higher singlet states as well as extended triplet lifetimes. However, Bz shows the highest initiation efficiency for polymerization of MMA at a wavelength of 351 nm compared to both MMMP (Bz:MMMP corresponds to 1:0.63) and 4MB (Bz:4MB corresponds to 1:0.86). The current study thus evidences that the combination of PLP-ESI-MS and TA experiments allows for arriving at quantitative initiation abilities of identical radical fragments originating from disparate source molecules. However, the quantitative initiation evaluation of different fragments originating from disparate source molecules requires additional information regarding the fragments' reactivity towards vinyl bonds.
Co-reporter:Johannes Willenbacher;Ozcan Altintas;Peter W. Roesky
Macromolecular Rapid Communications 2014 Volume 35( Issue 1) pp:45-51
Publication Date(Web):
DOI:10.1002/marc.201300594
Co-reporter:Thomas Tischer;Tanja K. Claus;Kim K. Oehlenschlaeger;Vanessa Trouillet;Michael Bruns;Alexer Welle;Katharina Linkert;Anja S. Goldmann;Hans G. Börner
Macromolecular Rapid Communications 2014 Volume 35( Issue 12) pp:1121-1127
Publication Date(Web):
DOI:10.1002/marc.201400088
Co-reporter:Florian Szillat;Bernhard V. K. J. Schmidt;Artur Hubert;Helmut Ritter
Macromolecular Rapid Communications 2014 Volume 35( Issue 14) pp:1293-1300
Publication Date(Web):
DOI:10.1002/marc.201400122
Co-reporter:Alexer P. Haehnel;Benjamin Wenn;Katrin Kockler;Tobias Bantle;Andrea M. Misske;Friederike Fleischhaker;Thomas Junkers
Macromolecular Rapid Communications 2014 Volume 35( Issue 23) pp:2029-2037
Publication Date(Web):
DOI:10.1002/marc.201400479
Co-reporter:Kayte Ranieri;Guillaume Delaittre;Thomas Junkers
Macromolecular Rapid Communications 2014 Volume 35( Issue 23) pp:2023-2028
Publication Date(Web):
DOI:10.1002/marc.201400518
Co-reporter:Bernhard V. K. J. Schmidt;Johannes Elbert;Markus Gallei
Macromolecular Rapid Communications 2014 Volume 35( Issue 7) pp:708-714
Publication Date(Web):
DOI:10.1002/marc.201300870
Co-reporter:Kayte Ranieri;Joke Venbergh;Thomas Junkers
Macromolecular Chemistry and Physics 2014 Volume 215( Issue 20) pp:1991-2000
Publication Date(Web):
DOI:10.1002/macp.201300740
Co-reporter:Dr. Bernhard V. K. J. Schmidt;Dr. Christopher Barner-Kowollik
ChemCatChem 2014 Volume 6( Issue 11) pp:3060-3062
Publication Date(Web):
DOI:10.1002/cctc.201402456
Co-reporter:Andrea Hufendiek, Vanessa Trouillet, Michael A. R. Meier, and Christopher Barner-Kowollik
Biomacromolecules 2014 Volume 15(Issue 7) pp:
Publication Date(Web):May 15, 2014
DOI:10.1021/bm500416m
Well-defined cellulose-graft-polyacrylamide copolymers were synthesized in a grafting-from approach by reversible addition–fragmentation chain transfer polymerization (RAFT). A chlorine moiety (degree of substitution DS(Cl) ≈ 1.0) was introduced into the cellulose using 1-butyl-3-methylimidazolium chloride (BMIMCl) as solvent before being substituted by a trithiocarbonate moiety resulting in cellulose macro-chain transfer agents (cellulose-CTA) with DS(RAFT) of 0.26 and 0.41. Poly(N,N-diethylacrylamide) (PDEAAm) and poly(N-isopropylacrylamide) (PNIPAM) were subsequently grafted from these cellulose-CTAs and the polymerization kinetics, the molecular weight characteristics and the product composition were studied by nuclear magnetic resonance spectroscopy, X-ray photoelectron spectroscopy, and size exclusion chromatography of the polyacrylamides after cleavage from the cellulose chains. The number-average molecular weights, Mn, of the cleaved polymers ranged from 1100 to 1600 g mol–1 for PDEAAm (dispersity Đ = 1.4–1.8) and from 1200 to 2600 g mol –1 for PNIPAM (Đ = 1.7–2.1). The LCST behavior of the cellulose-graft-copolymers was studied via the determination of cloud point temperatures, evidencing that the thermoresponsive properties of the hybrid materials could be finely tuned between 18 and 26 °C for PDEAAm and between 22 and 26 °C for PNIPAM side chains.
Co-reporter:Lebohang Hlalele, Dagmar R. D’hooge, Christoph J. Dürr, Andreas Kaiser, Sven Brandau, and Christopher Barner-Kowollik
Macromolecules 2014 Volume 47(Issue 9) pp:2820-2829
Publication Date(Web):April 23, 2014
DOI:10.1021/ma500055q
The successful RAFT-mediated ab initio emulsion copolymerization of acrylonitrile and 1,3-butadiene using 2-(((dodecylsulfanyl)carbonothioyl)sulfanyl)propanoic acid (DoPAT) is reported at 45–55 °C. The number-average molecular weight exhibits a linear evolution as a function of monomer conversion (5000 ≤ Mn (g mol–1) ≤ 41 000, 1.3 ≤ Đ (−) ≤ 3.3). Relatively good control (e.g., Đ ≈ 1.2 for selected conditions) over the polymerization up to moderate monomer conversion (50–60%) was attained when the employed initial molar ratio of RAFT agent to initiator was 2.5 or higher. Good ω-end-group functionality is evidenced by chain extension of NBR with a polystyrene block, with both 1H NMR and SEC showing the average fraction of the NBR block as ca. 75 mol%. A kinetic model implemented via the PREDICI software package confirms the experimental findings, including a semiempirical approach to account for branch formation. The onset of the loss in control over the copolymerization at conversions >40% was tentatively attributed to branch formation. The current study evidences that RAFT mediated ab initio emulsion polymerization of 1,3-butadiene and acrylonitrile is a viable polymerization protocol for the synthesis of well-defined next generation nitrile–butadiene rubbers including in industrial context.
Co-reporter:Alexander P. Haehnel, Maria Schneider-Baumann, Lukas Arens, Andrea M. Misske, Friederike Fleischhaker, and Christopher Barner-Kowollik
Macromolecules 2014 Volume 47(Issue 10) pp:3483-3496
Publication Date(Web):May 5, 2014
DOI:10.1021/ma500304f
The Arrhenius parameters of the propagation rate coefficient, kp, are determined via the IUPAC recommended pulsed laser polymerization–size exclusion chromatography (PLP-SEC) method for two linear alkyl acrylates (stearyl and behenyl acrylate), four branched alkyl acrylates (isononyl (INA-A), tridecyl (TDN-A and TDA-A), and henicosyl acrylate (C21A)), and two branched alkyl methacrylates (tridecyl methacrylates (TDN-MA and TDA-MA)) in bulk. Furthermore, the above stated acrylates and heptadecyl acrylate (C17A) were studied in 1 M solution in butyl acetate (BuAc). On the basis of such a wide data basis in combination with the already literature known data of relatives of the herein investigated monomers, we are able to identify and extend global trends and family type behavior for the propagation rate coefficients of a wide array of alkyl (meth)acrylates. In order to ensure a valid SEC evaluation, the polymer specific Mark–Houwnik–Kuhn–Sakurada (MHKS) parameters are determined for each of the polymers, via multidetector SEC analysis (multi angle laser light scattering (MALLS) in combination with differential viscosimetry (Visco) and refractive index (RI)) of narrowly distributed polymer samples obtained via fraction with a preparative SEC column. By employing further physicochemical polymer specific data (e.g., glass transition temperatures (Tg)), we provide a hypothesis for the reported trends and family type behaviors: (i) the steady increase of kp with increasing ester side chain length for linear alkyl (meth)acrylates may be explained by a decreasing concentration of the polar ester moieties, resulting in a decreasing stabilization of the attacking radical in the transition state of the propagation reaction, and (ii) the family type behavior of the branched alkyl methacrylates can be understood by considering steric and entropic influences. For the branched alkyl acrylates, no clear trend is detectable, and a family type behavior is clearly not observed in contrast to the corresponding methacrylates.
Co-reporter:Eva Blasco, Bernhard V. K. J. Schmidt, Christopher Barner-Kowollik, Milagros Piñol, and Luis Oriol
Macromolecules 2014 Volume 47(Issue 11) pp:3693-3700
Publication Date(Web):May 21, 2014
DOI:10.1021/ma500254p
We report the synthesis and characterization of a novel azobenzene-containing miktoarm star polymer AB3 (Mn = 9700 g mol–1, ĐM = 1.10) as well as its self-assembly properties in water. The miktoarm copolymer is composed of a hydrophobic azopolymer and three hydrophilic PEG arms (Mn = 600 g mol–1). The hydrophobic/hydrophilic ratio of the amphiphilic miktoarm polymer is 78/22, leading to the formation of stable polymeric vesicles in water evidenced via TEM and cryo-TEM imaging. The photoresponse of these vesicles has been investigated by irradiation with UV light (λ = 350–400 nm) causing the disruption of the self-assemblies. Encapsulation of both hydrophilic and hydrophobic fluorescent probes, i.e., Nile Red and Rhodamine B, and the use of light as an external stimulus to trigger the release of the probes have also been demonstrated.
Co-reporter:Ozcan Altintas, Peter Krolla-Sidenstein, Hartmut Gliemann, and Christopher Barner-Kowollik
Macromolecules 2014 Volume 47(Issue 17) pp:5877-5888
Publication Date(Web):August 19, 2014
DOI:10.1021/ma501186k
We report the precision single-chain folding of narrow dispersity diblock copolymers via pairwise orthogonal multiple hydrogen bonding motifs and single chain selected point folding. Well-defined linear polystyrene (PS) and poly(n-butyl acrylate) (PnBA) carrying complementary recognition units have been synthesized via activators regenerated by electron transfer/atom transfer radical polymerization (ARGET ATRP) utilizing functional initiators yielding molecular weights of Mn,SEC = 10900 Da, Đ = 1.09 and Mn,SEC = 3900 Da, Đ = 1.10, respectively. The orthogonal hydrogen bonding recognition motifs were incorporated into the polymer chain ends of the respective building blocks (to yield an eight shaped single chain folded polymers). Diblock copolymer formation was achieved via the Cu(I) catalyzed azide–alkyne cycloaddition (CuAAC) reaction, while the single-chain folding of the prepared linear diblock copolymer–at low concentrations–was driven by orthogonal multiple hydrogen bonds via three-point thymine–diaminopyridine and six-point cyanuric acid–Hamilton wedge self-association. The self-folding process was followed by proton nuclear magnetic resonance (1H NMR) spectroscopy focused on the respective recognition pairs at low temperature. In addition, the single-chain folding of the diblock copolymer was analyzed by dynamic light scattering (DLS) and concentration dependent diffusion ordered NMR spectroscopy (DOSY) as well as atomic force microscopy (AFM), providing a limiting concentration for self-folding (in dichloromethane at ambient temperature) of close to 10 mg mL–1.
Co-reporter:Cesar Rodriguez-Emmenegger;Corinna M. Preuss;Basit Yameen;Ognen Pop-Georgievski;Michael Bachmann;Jan O. Mueller;Michael Bruns;Anja S. Goldmann;Martin Bastmeyer
Advanced Materials 2013 Volume 25( Issue 42) pp:6123-6127
Publication Date(Web):
DOI:10.1002/adma.201302492
Co-reporter:Benjamin Richter;Thomas Pauloehrl;Johannes Kaschke;Dagmar Fichtner;Joachim Fischer;Alexra M. Greiner;Doris Wedlich;Martin Wegener;Guillaume Delaittre;Martin Bastmeyer
Advanced Materials 2013 Volume 25( Issue 42) pp:6117-6122
Publication Date(Web):
DOI:10.1002/adma.201302678
Co-reporter:Eva Blasco;Milagros Piñol;Luis Oriol;Bernhard V. K. J. Schmidt;Alexer Welle;Vanessa Trouillet;Michael Bruns
Advanced Functional Materials 2013 Volume 23( Issue 32) pp:4011-4019
Publication Date(Web):
DOI:10.1002/adfm.201203602
Abstract
The preparation of patterned photoswitchable surfaces by employing the nitrile imine-mediated tetrazole ene cycloaddition (NITEC) photoinduced reaction in the presence of dipolarophiles based on photoresponsive azobenzene moieties is reported. The dipolarophile used is a maleimide carrying either an azobenzene unit or a first generation dendron containing two azobenzene units. X-ray photoelectron spectroscopy (XPS) is employed to analyze the functionalized silicon wafers, while time-of-flight secondary ion mass spectrometry (ToF-SIMS) evidences the spatial control of the functionalization of the surface achieved by using a micropatterned shadow mask. Water contact angle measurements and optical inspection observing the behavior of a water droplet demonstrate the photoinduced change on wettability of the structured functionalized surfaces due to the reversible trans-to-cis isomerization of the azobenzene moities.
Co-reporter:Nathalie K. Guimard, Junming Ho, Josef Brandt, Ching Yeh Lin, Mansoor Namazian, Jan O. Mueller, Kim K. Oehlenschlaeger, Stefan Hilf, Albena Lederer, Friedrich G. Schmidt, Michelle L. Coote and Christopher Barner-Kowollik
Chemical Science 2013 vol. 4(Issue 7) pp:2752-2759
Publication Date(Web):02 May 2013
DOI:10.1039/C3SC50642H
The widely accepted approach for controlling polymer debonding/rebonding properties in responsive materials has been to purposefully engineer the functional end-groups responsible for monomer dynamic bonding. Here, however, we evidence that the debonding temperature of a polymer can also be tuned by changing the chain length of the polymer building blocks, thus altering the entropy released on debonding. Entropy driven debonding, as governed by building block chain length, is suggested theoretically and realized experimentally for two Diels–Alder polymer systems, each based on a different difunctional diene and a common difunctional dienophile. In each case a significant decrease (as much as 60 °C) in the retro Diels–Alder temperature was observed when the chain length of the difunctional dienophile building block was increased. These results have the potential to fundamentally change the approach utilized to design materials capable of bonding reversibly on demand.
Co-reporter:Thomas Pauloehrl, Alexander Welle, Kim K. Oehlenschlaeger and Christopher Barner-Kowollik
Chemical Science 2013 vol. 4(Issue 9) pp:3503-3507
Publication Date(Web):17 Jun 2013
DOI:10.1039/C3SC50815C
A novel and very simple photochemical strategy that utilizes light as a facile means to provide spatio-temporal control for the direct covalent immobilization of nucleophiles is presented. The concept is based upon the efficient trapping of photogenerated thioaldehydes by amines, hydroxylamines, and thiols. Surface patterns of polymers and small molecules bearing pendant amine-, hydroxylamine- or thiol moieties were successfully generated and imaged in a time-of-flight secondary-ion mass spectrometry (ToF-SIMS) investigation.
Co-reporter:Mirela Zamfir, Cesar Rodriguez-Emmenegger, Stella Bauer, Leonie Barner, Axel Rosenhahn and Christopher Barner-Kowollik
Journal of Materials Chemistry A 2013 vol. 1(Issue 44) pp:6027-6034
Publication Date(Web):19 Aug 2013
DOI:10.1039/C3TB20880J
The reversible addition–fragmentation chain transfer polymerization of 2-hydroxyethyl methacrylate (HEMA) from surfaces (S-RAFT) using an R-group-attached chain transfer agent (CTA) is presented. The approach was exploited for the efficient preparation of well-defined PHEMA brushes of up to 50 nm thickness in a controlled fashion without using any cytotoxic catalyst. The chemical composition, morphology and wettability of the samples were assessed by X-ray photoelectron spectroscopy, atomic force microscopy and water contact angle measurements, while the growth kinetics were studied by monitoring the dry thickness via spectroscopic ellipsometry. The mechanism and kinetics of the RAFT polymerization on the surface – in the presence of a sacrificial CTA and of solvent mixtures with different polarities – were investigated. A marked effect of the concentration of the sacrificial CTA on the kinetics was observed. Importantly – and for the first time – the living PHEMA brushes were exploited as macroRAFT agents for chain extension, and thicknesses up to 70 nm were achieved. The prepared PHEMA brushes were challenged with protein solutions demonstrating their resistance to fouling.
Co-reporter:Dennis M. Bauer, Anita Rogge, Lukas Stolzer, Christopher Barner-Kowollik and Ljiljana Fruk
Chemical Communications 2013 vol. 49(Issue 77) pp:8626-8628
Publication Date(Web):31 Jul 2013
DOI:10.1039/C3CC43291B
DNA was modified with a photo-reactive caged diene allowing for the modification of dienophile containing proteins under mild irradiation conditions to afford fully functional DNA–protein conjugates.
Co-reporter:Basit Yameen, Cesar Rodriguez-Emmenegger, Corinna M. Preuss, Ognen Pop-Georgievski, Elisseos Verveniotis, Vanessa Trouillet, Bohuslav Rezek and Christopher Barner-Kowollik
Chemical Communications 2013 vol. 49(Issue 77) pp:8623-8625
Publication Date(Web):22 Jul 2013
DOI:10.1039/C3CC44683B
Cyclopentadienyl end-capped poly(3-hexylthiophene) was employed to fabricate conductive surface tethered polymer brushes via a facile route based on cyclopentadiene–maleimide Diels–Alder ligation. The efficient nature of the Diels–Alder ligation was further combined with a biomimetic polydopamine-assisted functionalization of surfaces, making it an access route of choice for P3HT surface immobilization.
Co-reporter:Basit Yameen, Cesar Rodriguez-Emmenegger, Ishtiaq Ahmed, Corinna M. Preuss, Christoph J. Dürr, Nicolas Zydziak, Vanessa Trouillet, Ljiljana Fruk and Christopher Barner-Kowollik
Chemical Communications 2013 vol. 49(Issue 60) pp:6734-6736
Publication Date(Web):04 Jun 2013
DOI:10.1039/C3CC43361G
An unprecedented one-pot procedure employing a cyclopentadienyl functionalized RAFT agent allowed the grafting of poly(carboxybetaine acrylamide) – a highly functional and biocompatible polymer – from the surface of pristine SWCNTs. The pendant carboxylic acid groups of the surface grafted polymer were further conjugated with single-stranded (ss)-DNA, which was successfully hybridized with a Cy5 labelled complementary DNA strand.
Co-reporter:Mathias Glassner, Kim K. Oehlenschlaeger, Alexander Welle, Michael Bruns and Christopher Barner-Kowollik
Chemical Communications 2013 vol. 49(Issue 6) pp:633-635
Publication Date(Web):23 Nov 2012
DOI:10.1039/C2CC37651B
An efficient method for polymer surface patterning via Diels–Alder trapping of photo-generated thioaldehydes is presented. It is demonstrated that thioaldehyde end-groups generated by photolysis of phenacyl sulfides can be quantitatively trapped with various dienes. Poly(ethylene glycol) is immobilized on a surface in a spatially controlled fashion via irradiation through a shadow mask.
Co-reporter:Christiane Lang, Kai Pahnke, Claude Kiefer, Anja S. Goldmann, Peter W. Roesky and Christopher Barner-Kowollik
Polymer Chemistry 2013 vol. 4(Issue 21) pp:5456-5462
Publication Date(Web):28 Jun 2013
DOI:10.1039/C3PY00648D
Polymers with palladium ligating moieties in the lateral chain were synthesized via consecutive CuAAC ligation. An in-depth catalyst screening was performed to optimize the reaction conditions. The obtained polymers, quantitatively loaded with palladium complexes, feature high molar masses of up to Mn = 30000 g mol−1. The complex formation is underpinned by model complex studies. 2,6-Bis(1-(p-tolyl)-1H-1,2,3-triazol-4-yl)pyridine was used as the model ligand. Reaction of the ligand with [Pd(cod)Cl2] resulted in the corresponding palladium model complex.
Co-reporter:Christoph J. Dürr, Lebohang Hlalele, Maria Schneider-Baumann, Andreas Kaiser, Sven Brandau and Christopher Barner-Kowollik
Polymer Chemistry 2013 vol. 4(Issue 17) pp:4755-4767
Publication Date(Web):14 Jun 2013
DOI:10.1039/C3PY00580A
The microstructure of acrylonitrile–butadiene rubber (NBR) was shown to be dependent on the polymerization conditions. The NBR investigated in the current study was obtained via radical copolymerization under azeotropic conditions (AN/BD = 38/62) in organic solution in the presence of either a conventional chain transfer agent or a reversible addition fragmentation chain transfer (RAFT) agent. The variation in the polymer microstructure was proven to originate from different radical environments during the polymerizations with initial radical initiator concentrations in the polymerizations studied ranging from 1.0 mM to 34.1 mM. The variation of the polymer microstructure was evidenced by triple SEC measurements, making use of the simultaneous determination of molecular weights with two independent methods, namely on-line viscometry and on-line light scattering. It is additionally evidenced that the microstructure shows a gradual variation during the course of a polymerization, a behaviour observed when the polymerizations were performed in the presence of an elevated initial radical initiator concentration. Furthermore, experimental evidence for the variation of the NBR microstructure during RAFT polymerizations is provided. At low conversions, a rather uniform polymer is obtained. With increasing conversion, a loss of the controlled character is observed and the microstructures approach those of nitrile rubber obtained by conventionally controlled free radical copolymerizations employing mercaptane transfer agents. Despite the differences in the polymer microstructure, it is possible to report a single set of MHKS parameters for the prepared NBR with azeotropic composition. A linear regression of the Mark–Houwink plots of the samples polymerized under different conditions gives values of K = (49.5 ± 5.5) × 10−5 dL g−1 and α = 0.689 ± 0.010 with a low error margin for SEC separation in THF at 25 °C. Weight average molecular weights of the investigated NBR samples were in the range of 40000 to 155000 g mol−1. The molecular weights of the copolymers determined via universal calibration with the MHKS parameters presented in the current study show a good agreement with molecular weights obtained from light scattering, underpinning the veracity of the obtained parameters.
Co-reporter:Kim K. Oehlenschlaeger, Nathalie K. Guimard, Josef Brandt, Jan O. Mueller, Ching Yeh Lin, Stefan Hilf, Albena Lederer, Michelle L. Coote, Friedrich G. Schmidt and Christopher Barner-Kowollik
Polymer Chemistry 2013 vol. 4(Issue 16) pp:4348-4355
Publication Date(Web):23 May 2013
DOI:10.1039/C3PY00476G
A new dithioester possessing a cyano Z-group (cyano-dithioester (CDTE)) has been synthesized via a 2-step, one-pot reaction. The cyano-substituted dithioester has been found to undergo fast reversible hetero-Diels–Alder (HDA) reactions at ambient temperature, without the need for a catalyst, as demonstrated by ESI-MS and UV-Vis experiments. To apply the bonding/debonding on demand system to materials science, a cyano-dithioester di-linker was synthesized and employed as a di-functional dienophile in a HDA-based polymerization reaction with a bis-cyclopentadiene polymer. The reversible bonding of the polymer systems were demonstrated by on-line UV-Vis spectroscopy, on-line NMR spectroscopy, and on-line high temperature DLS, as well as via GPC in situ trapping experiments and high-level ab initio molecular orbital calculations.
Co-reporter:Nicolas Zydziak, Basit Yameen and Christopher Barner-Kowollik
Polymer Chemistry 2013 vol. 4(Issue 15) pp:4072-4086
Publication Date(Web):13 Mar 2013
DOI:10.1039/C3PY00232B
To meet the ever growing demand for carbon nanomaterials with tailored properties, Diels–Alder reactions are emerging as an efficient alternative to other synthetic methods. From an application perspective, the development of convenient surface functionalization strategies for carbon nanostructures is of paramount importance. Pristine carbon nanostructures display a natural tendency to undergo Diels–Alder reactions with a range of functional dienes and dienophiles without the need of a catalyst. This has sparked significant scientific interest in exploiting the Diels–Alder reaction as a powerful strategy for their synthesis as well as for their subsequent surface functionalization. The present review highlights the remarkable role of Diels–Alder reactions for the synthesis of fullerenes, carbon nanotubes and graphene, and its promise as a facile carbon nanostructure functionalization strategy with small molecules and polymer chains. A critical overview of the recent developments evidencing the potential of Diels–Alder reactions as an efficient route to carbon based functional materials is presented.
Co-reporter:Joerdis Eisenblaetter, Michael Bruns, Ulrich Fehrenbacher, Leonie Barner and Christopher Barner-Kowollik
Polymer Chemistry 2013 vol. 4(Issue 8) pp:2406-2413
Publication Date(Web):08 Feb 2013
DOI:10.1039/C3PY00103B
Polystyrene-based copolymers synthesized by RAFT polymerization of 4-vinylbenzyl chloride (4-VBC) and styrene with subsequent azide transformation are used to prepare phosphorylated polymers via 1,3-dipolar cycloaddition with diphenyl prop-2-ynyl phosphoric ester (DPPP). To avoid metal salts in the reaction mixture of the 1,3-dipolar cycloaddition, a novel metal free approach was developed to synthesize DPPP. The successful conversion to phosphorylated polymers is confirmed by X-ray photoelectron spectroscopy (XPS), infrared (IR) spectroscopy as well as solid phase nuclear magnetic resonance (NMR) spectroscopy. Thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) are employed to determine the influence of the phosphoric ester on the thermal properties of the generated polymers. Through a series of TGA-MS measurements, the decomposition products of the phosphorylated polymers, e.g. styrene, phenyl, alkyne and diphenyl phosphite moieties, are determined.
Co-reporter:Nicolas Zydziak, Christof Hübner, Michael Bruns, Andrew P. Vogt and Christopher Barner-Kowollik
Polymer Chemistry 2013 vol. 4(Issue 5) pp:1525-1537
Publication Date(Web):26 Nov 2012
DOI:10.1039/C2PY20928D
Cyclopentadienyl end-capped poly(N-isopropylacrylamide) (PNIPAM-Cp, Mn = 5400 g mol−1, PDI = 1.13) was synthesized via a combination of RAFT (Reversible Addition–Fragmentation Chain Transfer) polymerization and modular conjugation (characterized via Nuclear Magnetic Resonance (NMR) as well as Electrospray Ionization-Mass Spectrometry (ESI-MS)), and reacted with untreated Single Walled Carbon Nanotubes (SWCNTs) as dienophiles in a Diels–Alder reaction with PNIPAM-Cp (diene) at ambient temperature in the absence of any catalyst. The obtained stimuli-responsive hybrid materials display thermo-responsive behaviour evidenced via UV-VIS-spectroscopy and Dynamic Light Scattering (DLS). The grafting density of the PNIPAM chains at the surface of the SWCNTs was determined via Thermogravimetric Analysis (TGA), Elemental Analysis (EA) and X-ray Photoelectron Spectroscopy (XPS), to be close to 0.0288 chains per nm2.
Co-reporter:Ozcan Altintas, David Schulze-Suenninghausen, Burkhard Luy, and Christopher Barner-Kowollik
ACS Macro Letters 2013 Volume 2(Issue 3) pp:211
Publication Date(Web):February 22, 2013
DOI:10.1021/mz400066r
A well-defined Hamilton wedge (HW) midchain functionalized block copolymer, i.e., polyethylene glycol-b-polystyrene (PEG-HW-PS, Mn,GPC = 5600 Da, PDI = 1.03), was successfully synthesized via a combination of atom transfer radical polymerization (ATRP) and copper-catalyzed azide alkyne cycloaddition (CuAAC). An α,ω-cyanuric acid (CA) difunctional linear homopolymer poly(n-butylacrylate) (CA-PnBA-CA, Mn,GPC = 8100 Da, PDI = 1.09) was concomitantly prepared via reversible addition–fragmentation chain transfer (RAFT) polymerization. Supramolecular H-shaped macromolecules were—for the first time—prepared through supramolecular self-assembly between HW and CA recognition motifs to generate (PS-b-PEG)·PnBA·(PS-b-PEG) and (PS-b-PS)·PnBA·(PS-b-PS) in CH2Cl2 or dichloromethane-d2 at ambient temperature. The self-assembly process (at a total concentration of the two species of close to 4.5 mM) was evidenced by proton nuclear magnetic resonance (1H NMR) spectroscopy, diffusion-ordered NMR spectroscopy (DOSY), and dynamic light scattering (DLS) analyses. The results derived via DOSY NMR experiments and DLS combined with a Job plot analysis and in-depth NMR titration experiments indicate that the formation of supramolecular H-shaped macromolecules in 2:1 stoichiometry is efficiently occurring via the employed complementary recognition motifs with high binding constants (between 1.2 and 1.5 × 105 L mol–1 at ambient temperature).
Co-reporter:Alexer S. Quick;Joachim Fischer;Benjamin Richter;Thomas Pauloehrl;Vanessa Trouillet;Martin Wegener
Macromolecular Rapid Communications 2013 Volume 34( Issue 4) pp:335-340
Publication Date(Web):
DOI:10.1002/marc.201200796
Abstract
Three-dimensional microstructures are fabricated employing the direct laser writing process and radical thiol-ene polymerization. The resin system consists of a two-photon photoinitiator and multifunctional thiols and olefins. Woodpile photonic crystals with 22 layers and a rod distance of 2 μm are fabricated. The structures are characterized via scanning electron microscopy and focused ion beam milling. The thiol-ene polymerization during fabrication is verified via infrared spectroscopy. The structures are grafted in a subsequent thiol-Michael addition reaction with different functional maleimides. The success of the grafting reaction is evaluated via laser scanning microscopy and X-ray photoelectron spectroscopy. The grafting density is calculated to be close to 200 molecules μm−2.
Co-reporter:Nicolas Zydziak;Corinna M. Preuss;Volker Winkler;Michael Bruns;Christof Hübner
Macromolecular Rapid Communications 2013 Volume 34( Issue 8) pp:672-680
Publication Date(Web):
DOI:10.1002/marc.201300025
Co-reporter:Anja S. Goldmann;Mathias Glassner;Andrew J. Inglis
Macromolecular Rapid Communications 2013 Volume 34( Issue 10) pp:810-849
Publication Date(Web):
DOI:10.1002/marc.201300017
Co-reporter:Andrew P. Vogt;Thomas Tischer;Udo Geckle;Alexra M. Greiner;Vanessa Trouillet;Michael Kaupp;Leonie Barner;Thorsten Hofe
Macromolecular Rapid Communications 2013 Volume 34( Issue 11) pp:916-921
Publication Date(Web):
DOI:10.1002/marc.201200834
Co-reporter:Bernhard V. K. J. Schmidt;Martin Hetzer;Helmut Ritter
Macromolecular Rapid Communications 2013 Volume 34( Issue 16) pp:1306-1311
Publication Date(Web):
DOI:10.1002/marc.201300478
Co-reporter:Corinna M. Preuss;Anja S. Goldmann;Vanessa Trouillet;Andreas Walther
Macromolecular Rapid Communications 2013 Volume 34( Issue 8) pp:640-644
Publication Date(Web):
DOI:10.1002/marc.201300094
Co-reporter:Francesca Bennet;Thomas Rölle;Thomas Fäcke;Marc-Stephan Weiser;Friedrich-Karl Bruder;Thomas Junkers
Macromolecular Chemistry and Physics 2013 Volume 214( Issue 2) pp:236-245
Publication Date(Web):
DOI:10.1002/macp.201200285
Abstract
The transfer reactions occurring during polymerization of 2-(phenylcarbamoyloxy)ethyl acrylate (PhCEA) were studied by a detailed product mapping with electrospray ionization mass spectrometry (ESI-MS). Unlike postulated before, PhCEA exhibits the same characteristic transfer reactions as other acrylic monomers at elevated temperatures, resulting in vinyl-terminated and saturated products. Transfer to monomer via abstraction of a hydrogen atom from the ester side chain as suggested before is not observed. When polymerized in a poly(ethylene glycol) (PEG) matrix to increase viscosity, transfer into matrix products are observed in minor amounts, demonstrating that PEG can be employed as a solvent to further increase the overall rate of polymerization without interference from side reactions.
Co-reporter:Alexer P. Haehnel;Sven Fleischmann;Pascal Hesse;Klaus-Dieter Hungenberg
Macromolecular Reaction Engineering 2013 Volume 7( Issue 1) pp:8-23
Publication Date(Web):
DOI:10.1002/mren.201200030
Co-reporter:Bernhard V. K. J. Schmidt, Martin Hetzer, Helmut Ritter, and Christopher Barner-Kowollik
Macromolecules 2013 Volume 46(Issue 3) pp:1054-1065
Publication Date(Web):February 1, 2013
DOI:10.1021/ma302386w
A novel triblock macromolecular architecture based on cyclodextrin (CD) complexation is presented. A CD-functionalized biocompatible poly(N-(2-hydroxypropyl)methacrylamide) (PHPMA) building block (3800 ≤ Mn ≤ 10 600 g mol–1; 1.29 ≤ ĐM ≤ 1.46) and doubly guest-containing poly(N,N-dimethylacrylamide) (PDMAAm) (6400 ≤ Mn ≤ 15 700 g mol–1; 1.06 ≤ ĐM ≤ 1.15) and poly(N,N-diethylacrylamide) (PDEAAm) (5400 ≤ Mn ≤ 12 100 g mol–1; 1.11 ≤ ĐM ≤ 1.33) segments were prepared via reversible addition–fragmentation chain transfer (RAFT) polymerization and subsequently utilized for the formation of a well-defined supramolecular ABA triblock copolymer. The block formation was evidenced via dynamic light scattering (DLS), nuclear Overhauser effect spectroscopy (NOESY), and turbidity measurements. Furthermore, the connection of the blocks was proven to be temperature responsive and—in the case of azobenzene guests—responsive to the irradiation with UV light. The application of these stimuli leads to the disassembly of the triblock copolymer, which was shown to be reversible. In the case of PDEAAm containing triblock copolymers, the temperature-induced aggregation was investigated as well.
Co-reporter:Martin Hetzer, Carolin Fleischmann, Bernhard V.K.J. Schmidt, Christopher Barner-Kowollik, Helmut Ritter
Polymer 2013 Volume 54(Issue 19) pp:5141-5147
Publication Date(Web):23 August 2013
DOI:10.1016/j.polymer.2013.07.031
Visual recognition of the formation of supramolecular graft polymers in aqueous solution driven by host-guest assembly is presented. The interaction between β-cyclodextrin (β-CD) and phenolphthalein moieties can be followed via a distinctive change of color in basic media by optical inspection. The phenolphthalein-containing lateral polymer chain was obtained via reversible addition-fragmentation chain transfer (RAFT) copolymerization of N,N-dimethylacrylamide and N-(2-hydroxy-5-(1-(4-hydroxyphenyl)-3-oxo-1,3-dihydroisobenzofuran-1-yl)benzyl)acrylamide. The RAFT polymerizations afforded statistical copolymers with molecular masses between 6900 and 8800 g mol−1 and molar mass dispersities (Đ) between 1.1 and 1.2. β-CD-functionalized building blocks were synthesized using a propargyl-functionalized chain transfer agent for the polymerization of N,N-diethylacrylamide followed by a copper(I)-catalyzed cycloaddition with β-CD-azide (Mn = 8200 g mol−1; Đ = 1.2). Self-assembly of both polymers to form supramolecular graft polymers was evidenced by 2D NOESY NMR, UV–vis spectroscopy and dynamic light scattering.
Co-reporter:Cedric Dommanget, Christophe Boisson, Bernadette Charleux, Franck D’Agosto, Vincent Monteil, Fernande Boisson, Thomas Junkers, Christopher Barner-Kowollik, Yohann Guillaneuf, and Didier Gigmes
Macromolecules 2013 Volume 46(Issue 1) pp:
Publication Date(Web):October 5, 2012
DOI:10.1021/ma3014806
Enhanced spin capturing polymerization (ESCP)—a recent and versatile technique in the field of controlled radical polymerization—achieves control over molecular weights and the synthesis of complex copolymer structures for a wide range of monomers. In the present work, the use of ESCP was extended to the radical polymerization of ethylene under mild conditions (low temperature and medium ethylene pressure) using a nitrone as spin trapping agent. It was demonstrated that the evolution of polyethylene (PE) molecular weight can be accurately described by classical ESCP kinetic equations. A PE bearing a midchain alkoxyamine function was thus obtained with high selectivity (90%). A more complex structure was produced from the radical polymerization of ethylene in the presence of a midchain alkoxyamine-functionalized polystyrene (PS) synthesized by ESCP in the form of ABA triblock copolymer (where A is polystyrene and B polyethylene).
Co-reporter:Martin Hetzer;Bernhard V. K. J. Schmidt;Helmut Ritter
Journal of Polymer Science Part A: Polymer Chemistry 2013 Volume 51( Issue 11) pp:2504-2517
Publication Date(Web):
DOI:10.1002/pola.26644
ABSTRACT
The design and synthesis of a new hydrophobic monomer, that is, 4-(tert-butyl)phenyl 6-acrylamidohexanoate (TBP-AA-HO) and its ability to form supramolecular host/guest complexes with β-cyclodextrin (CD) is described. The aqueous CD-mediated reversible addition fragmentation chain transfer (RAFT) polymerization affords molecular masses up to 8600 g mol−1 with polydispersities between 1.2 and 1.4. The surprisingly low molecular weights for higher monomer/chain transfer agent (CTA) ratios are investigated by comparing results obtained from free radical and RAFT radical polymerization in aqueous and organic media. The results indicate a steric hindrance caused by attached CD molecules on the growing polymer chain leading to stagnation of the polymerization process due to a restricted accessibility of the reactive chain end. This hypothesis is supported by matrix-assisted laser desorption/ionization time of flight mass spectrometry. Furthermore, the CD-mediated synthesis of amphiphilic diblock copolymers in variable aqueous media is described. Hydrophilic poly(N,N-dimethylacrylamide) macro-CTAs with different molecular weights are used to polymerize TBP-AA-HO at 50 °C. The diblock copolymers are analyzed by 1H-nuclear magnetic resonance spectroscopy and size exclusion chromatography. The results confirm the polymer structure and reveal similar limitations of chain growth as observed for the CD-mediated homopolymerization with a limit of 7000 g mol−1 for efficient chain extension. © 2013 Wiley Periodicals, Inc. J. Polym. Sci., Part A: Polym. Chem. 2013, 51, 2504–2517
Co-reporter:Christoph J. Dürr, Paul Lederhose, Lebohang Hlalele, Doris Abt, Andreas Kaiser, Sven Brandau, and Christopher Barner-Kowollik
Macromolecules 2013 Volume 46(Issue 15) pp:5915-5923
Publication Date(Web):July 29, 2013
DOI:10.1021/ma401154k
A highly selective photo-induced nitrile imine mediated tetrazole–ene coupling (NITEC) of chain-end-functionalized nitrile–butadiene rubber (NBR) is reported, providing nitrile rubbers with molar masses of up to 48 000 g·mol–1. NBR was obtained via the reversible addition–fragmentation chain transfer (RAFT) mediated copolymerization of acrylonitrile and 1,3-butadiene employing a novel photoreactive tetrazole-functionalized trithiocarbonate. The herein reported tetrazole-functionalized trithiocarbonate represents—to the best of our knowledge—the first ever reported photoreactive RAFT agent capable of undergoing light-induced ligations with enes. Molar masses of the tetrazole-functionalized NBRs were in the range of 1000 to 38 000 g·mol–1 with dispersities between 1.1 to 1.6. By an appropriate choice of the tetrazole substituents, a reaction of the in situ formed enophile with the double bonds or the nitrile moieties of the incorporated monomer units within the polymer backbone—present in high excess relative to the dipolarophile linker molecule—was not observed. Underpinned by DFT calculations, the selectivity was identified to originate from a reduced LUMO energy level of the maleimide linker compared to the nonactivated backbone olefins when employing a nitrile–imine of moderate reactivity.
Co-reporter:Thomas Tischer, Tanja K. Claus, Michael Bruns, Vanessa Trouillet, Katharina Linkert, Cesar Rodriguez-Emmenegger, Anja S. Goldmann, Sébastien Perrier, Hans G. Börner, and Christopher Barner-Kowollik
Biomacromolecules 2013 Volume 14(Issue 12) pp:
Publication Date(Web):October 15, 2013
DOI:10.1021/bm401274v
An efficient phototriggered Diels–Alder conjugation is utilized to graft in an effective and straightforward approach poly(trifluoro ethyl methacrylate) (Mn = 3700 Da, Đ = 1.27) and a model peptide (GIGKFLHS) onto thin hyaluronan films and cellulose surfaces. The surfaces were functionalized with an o-quinodimethane moiety – capable of releasing a caged diene – via carbodiimide mediated coupling. The o-quinodimethane group is employed as a photoactive linker to tether predefined peptide/polymer strands in a spatially controlled manner onto the biosurface by photoenol ligation. An in-depth characterization employing XPS, ToF-SIMS, SPR, ellipsometry, and AFM was conducted to evidence the effectiveness of the presented approach.
Co-reporter:Michael Kaupp, Thomas Tischer, Astrid F. Hirschbiel, Andrew P. Vogt, Udo Geckle, Vanessa Trouillet, Thorsten Hofe, Martina H. Stenzel, and Christopher Barner-Kowollik
Macromolecules 2013 Volume 46(Issue 17) pp:
Publication Date(Web):August 20, 2013
DOI:10.1021/ma401242g
The current contribution describes the combination of an efficient reversible deactivation radical polymerization process (reversible addition–fragmentation chain transfer (RAFT) polymerization) with a mild light-induced modular ligation technique. A novel RAFT-agent was synthesized which carries a photoactive group based on ortho-quinodimethane (photo-enol) chemistry. The novel photoreactive RAFT-agent controls the polymerization of a wide range of monomers such as styrene, N,N-dimethylacrylamide and a protected glycomonomer (2-(2′,3′,4′,6′-tetra-O-acetyl-β-d-mannosyloxy)ethyl acrylate) with dispersities between 1.07 and 1.17 (3500 g·mol–1 ≤ Mn ≤ 10100 g·mol–1) and quantitative end-group functionalization. The photo-enol group reacts with dieneophiles under mild irradiation (λmax = 320 nm) at ambient conditions – so that the RAFT-group remains intact – and without any catalyst to form block copolymers in a matter of minutes. Furthermore, the RAFT-polymers can be photografted onto porous polymeric (poly(glycidyl methacrylate)) microspheres, after a one-step prefunctionalization with maleimide moieties. The successful photografting is evidenced by scanning electron microscopy (SEM), elemental analysis (EA), X-ray photoelectron spectroscopy (XPS) and high resolution attenuated total reflectance (ATR) FT-IR microscopy, which leads to qualitative as well as quantitative grafting data. The grafting densities obtained for polystyrene are close to 0.11 chains per nm2 (Mn = 3900 g·mol–1) and for poly(N,N-dimethylacrylamide) close to 0.12 chains per nm2 (Mn = 3500 g·mol–1). To highlight the additional benefit of employing a light-induced grafting reaction, Janus microspheres were prepared with poly(N,N-dimethylacrylamide) employing a Pickering emulsion approach and illustrated via ATR-FT-IR microscopy.
Co-reporter:Kim K. Oehlenschlaeger;Jan O. Mueller;Niklas B. Heine;Mathias Glassner;Dr. Nathalie K. Guimard;Dr. Guillaume Delaittre;Dr. Friedrich G. Schmidt;Dr. Christopher Barner-Kowollik
Angewandte Chemie 2013 Volume 125( Issue 2) pp:791-796
Publication Date(Web):
DOI:10.1002/ange.201206905
Co-reporter:Thomas Pauloehrl;Dr. Alexer Welle;Dr. Michael Bruns;Katharina Linkert;Dr. Hans G. Börner;Dr. Martin Bastmeyer;Dr. Guillaume Delaittre;Dr. Christopher Barner-Kowollik
Angewandte Chemie 2013 Volume 125( Issue 37) pp:9896-9900
Publication Date(Web):
DOI:10.1002/ange.201302040
Co-reporter:Kim K. Oehlenschlaeger;Jan O. Mueller;Niklas B. Heine;Mathias Glassner;Dr. Nathalie K. Guimard;Dr. Guillaume Delaittre;Dr. Friedrich G. Schmidt;Dr. Christopher Barner-Kowollik
Angewandte Chemie International Edition 2013 Volume 52( Issue 2) pp:762-766
Publication Date(Web):
DOI:10.1002/anie.201206905
Co-reporter:Thomas Pauloehrl;Dr. Alexer Welle;Dr. Michael Bruns;Katharina Linkert;Dr. Hans G. Börner;Dr. Martin Bastmeyer;Dr. Guillaume Delaittre;Dr. Christopher Barner-Kowollik
Angewandte Chemie International Edition 2013 Volume 52( Issue 37) pp:9714-9718
Publication Date(Web):
DOI:10.1002/anie.201302040
Co-reporter:Basit Yameen, Nicolas Zydziak, Steffen M. Weidner, Michael Bruns, and Christopher Barner-Kowollik
Macromolecules 2013 Volume 46(Issue 7) pp:2606-2615
Publication Date(Web):March 28, 2013
DOI:10.1021/ma4004055
The development of a facile covalent strategy for the fabrication of organic conducting polymers (OCPs)/carbon nanotubes (CNTs) based molecular hybrid materials remains a challenge and is expected to address the detrimental intrinsic bundling issue of CNTs. In view of the pristine CNTs’ ability to undergo Diels–Alder reactions with dienes, we report the synthesis of a novel poly(3-hexylthiophene) (P3HT) based organic conducting polymer (OCP) with terminal cyclopentadienyl (Cp) groups. The synthetic strategy employed is based on a combination of in situ end group functionalization via Grignard metathesis (GRIM) polymerization and a subsequent end group switching via reaction with nickelocene. Characterization data from Matrix-assisted laser desorption-ionization time-of-flight mass spectrometry (MALDI–TOF MS) fully support the successful synthesis of monofunctional Cp-capped P3HT, which was found to be highly reactive toward dienophile end-capped polystyrene (PS). The Cp-capped P3HT was subsequently ligated to the surface of pristine single walled CNTs (SWCNTs). The resulting P3HT/SWCNTs molecular hybrid material was characterized using thermogravimetric analysis (TGA), elemental analysis (EA), X-ray photoelectron spectroscopy (XPS), and high resolution transmission electron microscopy (HRTEM). The data from TGA, EA, and XPS were used to quantitatively deduce the grafting density. P3HT/SWCNTs prepared with Cp capped P3HT was found to contain 2 times more P3HT than the reference sample, featuring a grafting density of 0.0510 chains·nm–2 and a periodicity of 1 P3HT chain per 748 carbon atoms of the SWCNTs. HRTEM revealed individual SWCNTs wrapped with P3HT whereas in the reference sample P3HT was adsorbed on the bundles of the SWCNTs. The results presented here provide a new avenue for designing novel materials based on CNTs and OCPs.
Co-reporter:Susanne Hansson, Vanessa Trouillet, Thomas Tischer, Anja S. Goldmann, Anna Carlmark, Christopher Barner-Kowollik, and Eva Malmström
Biomacromolecules 2013 Volume 14(Issue 1) pp:
Publication Date(Web):October 8, 2012
DOI:10.1021/bm3013132
In the present study, the two grafting techniques grafting-from — by activators regenerated by electron transfer atom transfer radical polymerization (ARGET ATRP) — and grafting-to — by copper(I)-catalyzed azide–alkyne cycloaddition (CuAAC) — were systematically compared, employing cellulose as a substrate. In order to obtain a meaningful comparison, it is crucial that the graft lengths of the polymers that are grafted from and to the substrates are essentially identical. Herein, this was achieved by utilizing the free polymer formed in parallel to the grafting-from reaction as the polymer for the grafting-to reaction. Four graft lengths were investigated, and the molar masses of the four free polymers (21 ≤ Mn ≤ 100 kDa; 1.07 ≤ ĐM ≤ 1.26), i.e. the polymers subsequently employed in the grafting-to reaction, were shown to be in the same range as the molar masses of the polymers grafted from the surface (23 ≤ Mn ≤ 87 kDa; 1.08 ≤ ĐM ≤ 1.31). The molecular weights of the chains grafted from the surface were established after cleavage from the cellulose substrates via size exclusion chromatography (SEC). High-resolution Fourier transform infrared microscopy (FT-IRM) was employed as an efficient tool to study the spatial distribution of the polymer content on the grafted substrates. In addition, the functionalized substrates were analyzed by X-ray photoelectron spectroscopy (XPS), contact angle (CA) measurements, and field-emission scanning electron microscopy (FE-SEM). For cellulose substrates modified via the grafting-from approach, the content of polymer on the surfaces increased with increasing graft length, confirming the possibility to tailor not only the length of the polymer grafts but also the polymeric content on the surface. In comparison, for the grafting-to reaction, the grafted content could not be controlled by varying the length of the preformed polymer: the polymer content was essentially the same for the four graft lengths. Consequently, the obtained results, when employing cellulose as a substrate and under these conditions, suggest that the grafting-from approach is superior to the grafting-to technique with respect to controlling the distribution of the polymeric content on the surface.
Co-reporter:Ozcan Altintas, Kamran Riazi, Richmond Lee, Ching Y. Lin, Michelle L. Coote, Manfred Wilhelm, and Christopher Barner-Kowollik
Macromolecules 2013 Volume 46(Issue 20) pp:8079-8091
Publication Date(Web):October 7, 2013
DOI:10.1021/ma401749h
Although controlled/living radical polymerization processes have significantly facilitated the synthesis of well-defined low polydispersity polymers with specific functionalities, a detailed and systematic knowledge of the thermal stability of the products–highly important for most industrial processes–is not available. Linear polystyrene (PS) carrying a trithiocarbonate mid-chain functionality (thus emulating the structure of the Z-group approach via reversible addition–fragmentation chain transfer (RAFT) based macromolecular architectures) with various chain lengths (20 kDa ≤ Mn,SEC ≤ 150 kDa, 1.27 ≤ Đ = Mw/Mn ≤ 1.72) and chain-end functionality were synthesized via RAFT polymerization. The thermal stability behavior of the polymers was studied at temperatures ranging from 100 to 200 °C for up to 504 h (3 weeks). The thermally treated polymers were analyzed via size exclusion chromatography (SEC) to obtain the dependence of the polymer molecular weight distribution on time at a specific temperature under air or inert atmospheres. Cleavage rate coefficients of the mid-chain functional polymers in inert atmosphere were deduced as a function of temperature, resulting in activation parameters for two disparate Mn starting materials (Ea = 115 ± 4 kJ·mol–1, A = 0.85 × 109 ± 1 × 109 s–1, Mn,SEC = 21 kDa and Ea = 116 ± 4 kJ·mol–1, A = 6.24 × 109 ± 1 × 109 s–1, Mn,SEC = 102 kDa). Interestingly, the degradation proceeds significantly faster with increasing chain length, an observation possibly associated with entropic effects. The degradation mechanism was explored in detail via SEC–ESI–MS for acrylate based polymers and theoretical calculations suggesting a Chugaev-type cleavage process. Processing of the RAFT polymers via small scale extrusion as well as a rheological assessment at variable temperatures allowed a correlation of the processing conditions with the thermal degradation properties of the polystyrenes and polyacrylates in the melt.
Co-reporter:Ozcan Altintas, Johannes Willenbacher, Kilian N. R. Wuest, Kim K. Oehlenschlaeger, Peter Krolla-Sidenstein, Hartmut Gliemann, and Christopher Barner-Kowollik
Macromolecules 2013 Volume 46(Issue 20) pp:8092-8101
Publication Date(Web):October 11, 2013
DOI:10.1021/ma4015033
We present a new ambient temperature synthetic approach for the preparation of single-chain polymeric nanoparticles (SCNPs) under mild conditions using a UV-light-triggered Diels–Alder (DA) reaction for the intramolecular cross-linking of single polymer chains. Well-defined random copolymers with varying contents of styrene (S) and 4-chloromethylstyrene (CMS) were synthesized employing a nitroxide-mediated radical polymerization (NMP) initiator functionalized with a terminal alkyne moiety. Postpolymerization modification with 4-hydroxy-2,5-dimethylbenzophenone (DMBP) and an N-maleimide (Mal) derivative led to the functional linear precursor copolymers. The intramolecular cross-linking was performed by activating the DMBP groups via irradiation with UV light of 320 nm for 30 min in diluted solution (cPolymer = 0.017 mg mL–1). The ensuing DA reaction between the activated DMBP and the Mal groups resulted in well-defined single-chain polymeric nanoparticles. To control the size of the SCNPs, random copolymers with varying CMS contents (i.e., different functional group densities (FGD)) were employed for the single-chain collapse. Additionally, monotethered nanoparticles were prepared via the copper-catalyzed azide–alkyne cycloaddition between the alkyne bearing copolymer with the highest FGD and an azide-terminated poly(ethylene glycol) (PEG) prior to UV-induced cross-linking. The formation of SCNPs was followed by size exclusion chromatography (SEC), nuclear magnetic resonance (NMR) spectroscopy, dynamic light scattering (DLS), and atomic force microscopy (AFM).
Co-reporter:Antoine Debuigne, Marie Hurtgen, Christophe Detrembleur, Christine Jérôme, Christopher Barner-Kowollik, Thomas Junkers
Progress in Polymer Science 2012 Volume 37(Issue 7) pp:1004-1030
Publication Date(Web):July 2012
DOI:10.1016/j.progpolymsci.2012.01.003
The current review focuses on the relevance and practical benefit of interpolymer radical coupling methods. The latter are developing rapidly and constitute a perfectly complementary macromolecular engineering toolbox to the controlled radical polymerization techniques (CRP). Indeed, all structures formed by CRP are likely to be prone to radical coupling reactions, which multiply the available synthetic possibilities. Basically, the coupling systems can be divided in two main categories. The first one, including the atom transfer radical coupling (ATRC), silane radical atom abstraction (SRAA) and cobalt-mediated radical coupling (CMRC), relies on the recombination of macroradicals produced from a dormant species. The second one, including atom transfer nitroxide radical coupling (ATNRC), single electron transfer nitroxide radical coupling (SETNRC), enhanced spin capturing polymerization (ESCP) and nitrone/nitroso mediated radical coupling (NMRC), makes use of a radical scavenger in order to promote the conjugation of the polymer chains. More than a compilation of macromolecular engineering achievements, the present review additionally aims to emphasize the particularities, synthetic potential and present limitations of each system.
Co-reporter:Anja S. Goldmann, Leonie Barner, Michael Kaupp, Andrew P. Vogt, Christopher Barner-Kowollik
Progress in Polymer Science 2012 Volume 37(Issue 7) pp:975-984
Publication Date(Web):July 2012
DOI:10.1016/j.progpolymsci.2011.11.008
Modular ligation strategies for the functionalization of polymeric microspheres provide new perspectives for their applications in material science. In the current trend article we highlight variable synthetic procedures for generating functional microspheres via orthogonal modular conjugation chemistries. An overview of the different surface chemistries available is provided, followed by surface-sensitive characterization techniques relevant for the microparticles. Finally, we explore future trends in modular orthogonal modification approaches on microparticles and provide an outlook on the perspectives that the field of surface-modification of polymeric microparticles holds.
Co-reporter:Mathias Glassner ; Guillaume Delaittre ; Michael Kaupp ; James P. Blinco
Journal of the American Chemical Society 2012 Volume 134(Issue 17) pp:7274-7277
Publication Date(Web):April 17, 2012
DOI:10.1021/ja301762y
Tailor-made water-soluble macromolecules, including a glycopolymer, obtained by living/controlled RAFT-mediated polymerization are demonstrated to react in water with diene-functionalized poly(ethylene glycol)s without pre- or post-functionalization steps or the need for a catalyst at ambient temperature. As previously observed in organic solvents, hetero-Diels–Alder (HDA) conjugations reached quantitative conversion within minutes when cyclopentadienyl moieties were involved. However, while catalysts and elevated temperatures were previously necessary for open-chain diene conjugation, additive-free HDA cycloadditions occur in water within a few hours at ambient temperature. Experimental evidence for efficient conjugations is provided via unambiguous ESI-MS, UV/vis, NMR, and SEC data.
Co-reporter:Mathias Dietrich;Guillaume Delaittre;James P. Blinco;Andrew J. Inglis;Michael Bruns
Advanced Functional Materials 2012 Volume 22( Issue 2) pp:304-312
Publication Date(Web):
DOI:10.1002/adfm.201102068
Abstract
The nitrile imine-mediated tetrazole-ene cycloaddition reaction (NITEC) is introduced as a powerful and versatile conjugation tool to covalently ligate macromolecules onto variable (bio)surfaces. The NITEC approach is initiated by UV irradiation and proceeds rapidly at ambient temperature yielding a highly fluorescent linkage. Initially, the formation of block copolymers by the NITEC methodology is studied to evidence its efficacy as a macromolecular conjugation tool. The grafting of polymers onto inorganic (silicon) and bioorganic (cellulose) surfaces is subsequently carried out employing the optimized reaction conditions obtained from the macromolecular ligation experiments and evidenced by surface characterization techniques, including X-ray photoelectron spectroscopy and FT-IR microscopy. In addition, the patterned immobilization of variable polymer chains onto profluorescent cellulose is achieved through a simple masking process during the irradiation.
Co-reporter:Thomas Tischer;Anja S. Goldmann;Katharina Linkert;Vanessa Trouillet;Hans G. Börner
Advanced Functional Materials 2012 Volume 22( Issue 18) pp:3853-3864
Publication Date(Web):
DOI:10.1002/adfm.201200266
Abstract
Hetero Diels-Alder (HDA) cycloaddition – as an effective modular conjugation approach – is employed to graft thioamide endfunctional oligopeptides onto solid cyclopentadienyl (Cp) functional cellulose substrates generating cellulose-peptide hybrid materials. The highly reactive Cp moieties serve as diene functionality in the consecutive HDA reaction on the biosubstrate surface. Oligopeptides (i.e., the model peptide Gly-Gly-Arg-Phe-Pro-Trp-Trp-Gly and the antimicrobial peptide tritrpticin) are functionalized at their N-termini employing strongly electron deficient thiocarbonyl thio compounds resulting in biomacromolecules bearing a thioamide endgroup. The dienophile- functional peptides readily undergo HDA reactions at ambient temperature and under mild conditions in solution with synthetic polymers as well as on solid (bio)substrates. An in-depth investigation is provided of the influence of the temperature, the Lewis acid catalysis and the side group exchange of thioamide functional oligopeptides reacting with Cp terminated poly(methyl methacrylate) (Mn = 2100 g·mol−1, PDI = 1.1) in homogenous solution as well as Cp functionalized cellulose in a heterogeneous system. To assess the success of the grafting reaction, the soluble samples were subjected to characterization methods such as size exclusion chromatography (SEC) and SEC-electrospray ionization mass spectrometry (SEC-ESI-MS). The heterogeneous “grafting-to” reactions were monitored using high resolution attenuated total reflection (ATR) Fourier transform infrared microscopy (HR-FTIRM) imaging, X-ray photoelectron spectroscopy (XPS) and elemental analysis. Evaluation via elemental analysis leads to quantitative peptide cellulose surface loading capacities.
Co-reporter:Matthias Winkler, Lucas Montero de Espinosa, Christopher Barner-Kowollik and Michael A. R. Meier
Chemical Science 2012 vol. 3(Issue 8) pp:2607-2615
Publication Date(Web):16 May 2012
DOI:10.1039/C2SC20402A
Heck coupling reactions are introduced as a modular, very efficient and highly orthogonal method for polymer–polymer conjugation. Several diblock and triblock copolymers (5200 Da ≤ Mn ≤ 17300 Da, 1.08 ≤ PDI ≤ 1.33) were prepared via Heck coupling reactions of acrylate-terminated and aryliodide-terminated polymers. The coupling reactions were performed using the so-called Jeffery's conditions, which allowed the use of equimolar amounts of reacting polymers and low reaction temperatures. Acrylated poly(ethylene glycol) monomethyl ether (PEG), poly(ε-caprolactone) (PCL) and polymers synthesized via head-to-tail selective acyclic diene metathesis (ADMET) polymerisation have been successfully conjugated with both a PEG and PCL containing an aryliodide moiety.
Co-reporter:Daniel Volz, Thomas Baumann, Harald Flügge, Mathias Mydlak, Tobias Grab, Michael Bächle, Christopher Barner-Kowollik and Stefan Bräse
Journal of Materials Chemistry A 2012 vol. 22(Issue 38) pp:20786-20790
Publication Date(Web):03 Sep 2012
DOI:10.1039/C2JM33291D
The production of solution-processed OLEDs requires materials suitable for subsequent multilayer deposition. In the current study, we present an autocatalytic method to crosslink a luminescent copper(I)-complex with a polymeric backbone, in which the emitter itself acts as catalyst. In a showcase reaction demonstrating this concept for the first time, we combined a highly luminescent binuclear copper(I)-complex with a polystyrene derivative in order to prove the potential of the protocol. The luminescence properties were only slightly affected by the crosslinking, while the general stability increased drastically, as proven by thermogravimetric analysis (TGA). OLED tests confirmed the fundamental suitability of the concept for device applications as well as for subsequent solution-based multilayer deposition.
Co-reporter:Bernhard V. K. J. Schmidt, Tobias Rudolph, Martin Hetzer, Helmut Ritter, Felix H. Schacher and Christopher Barner-Kowollik
Polymer Chemistry 2012 vol. 3(Issue 11) pp:3139-3145
Publication Date(Web):25 May 2012
DOI:10.1039/C2PY20293J
Three-armed star polymers with a cyclodextrin core and polyacrylamide arms were prepared via inclusion complex formation. A novel adamantyl-functionalized chain transfer agent was utilized in the reversible addition–fragmentation chain transfer (RAFT) polymerization of N,N-dimethylacrylamide and N,N-diethylacrylamide to afford guest-functionalized polymers. In combination with a three-pronged β-cyclodextrin core, supramolecular self-assembly was exploited for the formation of star-shaped architectures. This was demonstrated via a combination of dynamic light scattering (DLS) and 2D rotating frame nuclear Overhauser effect spectroscopy (ROESY) in D2O. Furthermore, turbidity measurements in the case of PDEAAm showed a rise in the LCST of the system. The complex formation was shown to be reversible, as DLS at elevated temperatures (70 °C) indicated a complete disassembly of the star polymers.
Co-reporter:Bernhard V. K. J. Schmidt, Martin Hetzer, Helmut Ritter and Christopher Barner-Kowollik
Polymer Chemistry 2012 vol. 3(Issue 11) pp:3064-3067
Publication Date(Web):13 Jun 2012
DOI:10.1039/C2PY20214J
We report the formation of a novel class of supramolecular miktoarm star polymers via cyclodextrin host–guest chemistry. Reversible addition–fragmentation chain transfer polymerization from difunctional guest- or alkyne-containing chain transfer agents and copper(I)-catalyzed azide–alkyne cycloaddition leads to host- and guest-containing acrylamido polymers, namely poly(N,N-dimethylacrylamide) and poly(N,N-diethylacrylamide). Subsequently, a supramolecular miktoarm star polymer was formed and characterized via dynamic light scattering and rotating frame nuclear Overhauser effect spectroscopy.
Co-reporter:Michael Kaupp, Andrew P. Vogt, Jens C. Natterodt, Vanessa Trouillet, Till Gruendling, Thorsten Hofe, Leonie Barner and Christopher Barner-Kowollik
Polymer Chemistry 2012 vol. 3(Issue 9) pp:2605-2614
Publication Date(Web):13 Jun 2012
DOI:10.1039/C2PY20369C
The facile and efficient functionalization of porous poly(glycidyl methacrylate) (pGMA) microspheres via hetero Diels–Alder (HDA) chemistry with poly(3-O-acryloyl-1,2:5,6-di-O-isopropylidene-α-D-glucofuranoside) (pAIpGlc) prepared by reversible addition–fragmentation chain transfer (RAFT) polymerization employing electron deficient thiocarbonylthio compounds (benzyl pyridin-2-yldithioformate (BPDF)) is described in detail. The efficiency of the employed ‘grafting to’ approach is qualitatively and quantitatively analyzed. Initially the microspheres are functionalized with a highly reactive diene – cyclopentadiene (Cp) – in one step with sodium cyclopentadienide, and subsequently reacted with a protected glycopolymer (number-average molecular weight, Mn = 4200 g mol−1; polydispersity index, PDI = 1.2) that carries a thiocarbonyl moiety functioning as a dienophile. The functionalization of the microspheres is achieved under mild conditions (T = 50 °C) with trifluoroacetic acid (TFA) as a readily removable catalyst. Deprotection of the grafted pAIpGlc to poly(3-O-acryloyl-α,β-D-glucopyranoside) (pAGlc) can be performed after functionalization in one pot with formic acid at ambient temperature. The obtained loading capacity is 2.63 × 1019 chains per g and the grafting density is close to 0.16 chains per nm2. Quantitative analysis of the grafting densities is achieved via elemental analysis; the pore size distribution before functionalization was analyzed by inverse size exclusion chromatography (iSEC). Further employed characterization techniques include scanning electron microscopy (SEM), X-ray photoelectron spectroscopy (XPS) and high resolution attenuated total reflectance (ATR) FT-IR microscopy supporting the successful modification of the microspheres.
Co-reporter:Christiane Lang, Claude Kiefer, Elise Lejeune, Anja S. Goldmann, Frank Breher, Peter W. Roesky and Christopher Barner-Kowollik
Polymer Chemistry 2012 vol. 3(Issue 9) pp:2413-2420
Publication Date(Web):22 Jun 2012
DOI:10.1039/C2PY20242E
Polystyrene based copolymers with palladium-ligating side groups were obtained by RAFT polymerization of chloromethyl styrene and styrene, subsequent azide transformation and quantitative copper-mediated 1,3-dipolar cycloaddition of ethynyl pyridine. Variable copolymer compositions were synthesized ranging from 10% to 50% with respect to the fraction of the palladium-ligating comonomer. The successful loading of the polymers with PdCl2 employing [Pd(COD)Cl2] (COD = 1,5-cyclooctadiene) as the metal source was confirmed by nuclear magnetic resonance spectroscopy (NMR) and elemental analysis. Absolute molecular weights of the Pd-containing polymers ranging from Mw = 16000 g mol−1 to 20000 g mol−1 were determined by employing static light scattering (SLS). In order to fully clarify the incorporation of palladium into the polymers, a PdCl2 complex of a chelating N donor ligand featuring the characteristic heterocyclic donor functions used to incorporate PdCl2 in the polymer was synthesized and characterized by single crystal X-ray diffraction and NMR analysis.
Co-reporter:Leonie Barner, Alexander S. Quick, Andrew P. Vogt, Volker Winkler, Thomas Junkers and Christopher Barner-Kowollik
Polymer Chemistry 2012 vol. 3(Issue 8) pp:2266-2276
Publication Date(Web):04 May 2012
DOI:10.1039/C2PY20272G
Complex cross-linked soluble architectures with molecular weights ranging from 25000 to 71000 g mol−1 and polydispersities (PDIs) ranging from 2.5 to 8.1 are generated using nitrone mediated chemistry. These cross-linked architectures are subsequently cleaved into their constituting fragments, i.e. linear chains with molecular weights ranging from 36000 to 40000 g mol−1 and a PDI of 2.0 or network fragments with molecular weights ranging from 28000 to 30000 g mol−1 and PDIs from 2.5 to 3.7, respectively. The cleavage into linear chains or network fragments depends on the pathway of network formation. Nitrone mediated reactions are also applied for the synthesis of microspheres. Conventional distillation precipitation polymerization was performed in the presence of a nitrone, leading to uniform microspheres with diameters ranging from 2.2 to 2.4 μm. These microspheres feature alkoxyamine functionalities throughout their interior and on their surface. The alkoxyamine functionality on the surface is employed in a subsequent nitroxide mediated polymerization (NMP) with N-isopropylacrylamide (NiPAAm) in a ‘grafting from’ approach to generate pNiPAAm on the surface of the microspheres. The resulting stimuli-responsive core–shell microspheres are characterized via SEM, XPS, elemental analysis as well as turbidity measurements. The characterization methods confirm the presence of a stimuli-responsive layer of pNiPAAm on the microspheres' surface.
Co-reporter:Thomas Pauloehrl, Guillaume Delaittre, Martin Bastmeyer and Christopher Barner-Kowollik
Polymer Chemistry 2012 vol. 3(Issue 7) pp:1740-1749
Publication Date(Web):20 Oct 2011
DOI:10.1039/C1PY00372K
A novel and efficient methodology for the light-triggered release of thiols at ambient temperature is presented, which can be utilized for the in situ modification of polymeric backbones prepared via radical polymerization. Initially, a model reaction on poly(ethylene glycol) methyl ether was examined via size-exclusion chromatography coupled with electrospray ionization-mass spectrometry (SEC/ESI-MS) to establish the photodeprotection feasibility of 2-nitrobenzyl thioether moieties in the presence of variable activators or catalysts employed are Michael-type or radical thiol–ene chemistries, respectively. When 0.01 eq. of dimethylphenylphosphine is employed, disulfide coupling is reduced to its minimum and quantitative phototriggered formation of thiol-capped poly(ethylene glycol) methyl ether species is observed after a 16 hour irradiation period at 320 nm by a low-cost light source. The concept is extended to polymer backbone modification by atom transfer radical polymerization of the novel photosensitive monomer: 2-((3-((2-nitrobenzyl)thio)propanoyl)oxy)ethyl methacrylate containing the 2-nitrobenzyl thioether moiety. Well-defined homopolymers (4700 g·mol−1 ≤ Mn ≤ 20000 g·mol−1, 1.29 ≤ PDI ≤ 1.40) containing one protected thiol per repeating unit were obtained and, upon a light stimulus (λmax = 320 nm), thiol entities are released along the lateral polymer chain. The photodeprotection process is mapped by exploiting the increased absorbance of photocleaved o-nitrosobenzaldehyde molecules at 345 nm and UV-Vis data suggests a quantitative backbone deprotection after a 16 hour irradiation time period. Further in situ functionalization of polymeric backbone is achieved via base-catalyzed maleimide–thiol addition at ambient temperature and its outcome is evidenced by a re-increased molecular weight in SEC, by virtue of decreased signal intensity of the 2-nitrobenzyl thioether moiety and the appearance of characteristic product protons in NMR spectroscopy (the polymer backbone functionalization is estimated as >90% by NMR analysis).
Co-reporter:Christoph J. Dürr, Sebastian G. J. Emmerling, Paul Lederhose, Andreas Kaiser, Sven Brandau, Michael Klimpel and Christopher Barner-Kowollik
Polymer Chemistry 2012 vol. 3(Issue 4) pp:1048-1060
Publication Date(Web):07 Feb 2012
DOI:10.1039/C2PY00547F
α-Functional nitrile butadiene rubber (NBR) building blocks were employed in the copper mediated 1,3-dipolar Huisgen coupling upon addition of 1,4-bis(azidomethyl)benzene (4). Polymer–polymer coupling afforded linear polymers with molecular weights ranging from 2500 g mol−1 to 97000 g mol−1 and polydispersities from 1.1 to 1.6. The α-functional NBR building blocks were obtained via the reversible addition–fragmentation chain transfer (RAFT) copolymerization of acrylonitrile (AN) and 1,3-butadiene (BD) at 100 °C, utilizing the high temperature azo initiator 1,1′-azobis(cyclohexane-1-carbonitrile) and chlorobenzene or acetone as solvents. A novel alkyne-functional trithiocarbonate 2 was synthesized in 64% yield via the N,N′-dicyclohexylcarbodiimide mediated coupling of 2-((dodecylsulfanyl)carbono-thioyl)sulfanyl propanoic acid (DoPAT, 1) and propargyl alcohol. 2 was shown to be an efficient controlling agent for the controlled/living radical copolymerization of acrylonitrile and 1,3-butadiene. The use of copper mediated azide–alkyne cycloaddition was extended towards the side-chain modification of acrylonitrile–butadiene rubbers as well as applied in the synthesis of branched and cross-linked NBR structures. For this purpose an acrylonitrile-1,3-butadiene–propargyl methacrylate (PMA) terpolymer of 3900 g mol−1 with a PDI of 1.3 was synthesized by a DoPAT-mediated RAFT polymerization. Herein, monomers were employed in the ratio of 56:35:9 (BD:AN:PMA). The ability of the terpolymer to undergo side-chain modification was demonstrated upon addition of 1-undecane azide. Cross-links were established via addition of 1,4-bis(azidomethyl)benzene. The current study provides the first successful approach to employ an orthogonal conjugation technique on this technically important class of synthetic rubbers.
Co-reporter:Dominik Voll;Andrea Hufendiek;Thomas Junkers
Macromolecular Rapid Communications 2012 Volume 33( Issue 1) pp:47-53
Publication Date(Web):
DOI:10.1002/marc.201100655
Abstract
Online size exclusion chromatography–electrospray ionization–mass spectrometry (SEC/ESI–MS) is employed for quantifying the overall initiation efficiencies of photolytically generated radical fragments. In a unique experiment, we present the first quantitative and systematic study of methyl-substituted acetophenone-type photoinitiators being employed in a single cocktail to initiate the free-radical polymerization of methyl methacrylate (MMA) in bulk. The photoinitiators are constituted of a set of two known and four new molecules, which represent an increasing number of methyl substituents on their benzoyl fragment, that is, benzoin, 4-methylbenzoin, 2,4-dimethylbenzoin, 2,4,6-trimethylbenzoin, 2,3,5,6-tetramethylbenzoin, and 2,3,4,5,6-pentamethylbenzoin. The absolute quantitative evaluation of the mass spectra shows a clear difference in the initiation ability of the differently substituted benzoyl-type radical fragments: Increasing the number of methyl substituents leads to a decrease in incorporation of the radical fragments.
Co-reporter:Thomas Junkers;Guillaume Delaittre;Robert Chapman;Fabian Günzler;Elena Chernikova
Macromolecular Rapid Communications 2012 Volume 33( Issue 11) pp:984-990
Publication Date(Web):
DOI:10.1002/marc.201200128
Abstract
A novel dithioester control agent [dimethyltetrathioterephtalate (DMTTT)] is presented for the thioketone-mediated radical polymerization (TKMP) of n-butyl acrylate. The rate of polymerization is significantly decreased in the presence of DMTTT indicating formation of dormant radical species. During polymerization, molar masses increase linearly with monomer conversion with reasonably narrow initial molar mass distributions (PDI between 1.3 and 1.8), whereas the dispersity increases during the course of the polymerization due to irreversible termination of both propagating and dormant radicals. The present results thus highlight the possibility of a mixed mechanism operating in RAFT polymerization, which combines slow fragmentation (long-lived intermediates) and intermediate radical termination.
Co-reporter:Ozcan Altintas
Macromolecular Rapid Communications 2012 Volume 33( Issue 11) pp:958-971
Publication Date(Web):
DOI:10.1002/marc.201200049
Abstract
The present feature article highlights the preparation of polymeric nanoparticles and initial attempts towards mimicking the structure of natural biomacromolecules by single chain folding of well-defined linear polymers through covalent and non-covalent interactions. Initially, the discussion focuses on the synthesis and characterization of single chain self-folded structures by non-covalent interactions. The second part of the article summarizes the folding of single chain polymers by means of covalent interactions into nanoparticle systems. The current state of the art in the field of single chain folding indicates that covalent-bond-driven nanoparticle preparation is well advanced, while the first encouraging steps towards building reversible single chain folding systems by the use of mutually orthogonal hydrogen-bonding motifs have been made.
Co-reporter:Andrew P. Vogt;Vanessa Trouillet;Alexra M. Greiner;Michael Kaupp;Udo Geckle;Leonie Barner;Thorsten Hofe
Macromolecular Rapid Communications 2012 Volume 33( Issue 13) pp:1108-1113
Publication Date(Web):
DOI:10.1002/marc.201200144
Abstract
Boronic acid-functionalized microspheres are prepared for the first time via mild epoxide ring opening based on porous cross-linked polymeric microspheres (diameter ≈ 10 μm, porosity ≈ 1000 Å). Quantitative chemical analysis by XPS and EA evidences that there is a greater functionalization with boronic acid when employing a sequential synthetic method [1.7 atom% boron (XPS); 1.12 wt% nitrogen (EA)] versus a one-pot synthetic method [0.2 atom% boron (XPS); 0.60 wt% nitrogen (EA)] yielding grafting densities ranging from approximately 2.5 molecules of boronic acid per nm2 to 1 molecule of boronic acid per nm2, respectively. Furthermore, the boronic acid-functionalized microspheres are conjugated with a novel fluorescent glucose molecule demonstrating a homogeneous spatial distribution of boronic acid.
Co-reporter:Guillaume Delaittre, Alexandra M. Greiner, Thomas Pauloehrl, Martin Bastmeyer and Christopher Barner-Kowollik
Soft Matter 2012 vol. 8(Issue 28) pp:7323-7347
Publication Date(Web):23 May 2012
DOI:10.1039/C2SM07407A
In the present review the principal strategies to chemically modify the surface of synthetic polymeric materials with small molecules for targeted cell adhesion are collated and critically discussed. The focus is purposely oriented on the chemistry involved in these modifications and neither the physical characterizations nor the activity evaluations resulting from these modifications are addressed in depth, although most reviewed examples demonstrate cell adhesion. Particularly, the introduction of a chemical anchor onto the polymeric substrate, the spacing via a linker between the polymer surface and the cell-binding motif, as well as the linkage generated on this cell-binding motif are discussed. Particular cases where variable substrate geometries or spatial patterning are achieved are additionally highlighted.
Co-reporter:Nathalie K. Guimard;Kim K. Oehlenschlaeger;Jiawen Zhou;Stefan Hilf;Friedrich Georg Schmidt
Macromolecular Chemistry and Physics 2012 Volume 213( Issue 2) pp:131-143
Publication Date(Web):
DOI:10.1002/macp.201100442
Abstract
The evolution of material design has mirrored advancements in the understanding of materials, nature, and the requirements of target applications. Originally, materials were only intended to play a passive role, but with a deepened understanding of material properties and design has come an improved ability to harness these properties to create materials with predetermined response mechanisms. This article has three aims: i) to briefly discuss the origin of and motivation for having materials that are capable of undergoing healing either extrinsically or intrinsically; ii) to present the most recent and promising advancements in the field of self-healing materials; and iii) to discuss important material design and property specifications that should be considered in order to promote the development of optimized self-healing materials.
Co-reporter:Christopher Barner-Kowollik;Friedrich Georg Schmidt
Macromolecular Chemistry and Physics 2012 Volume 213( Issue 2) pp:129-130
Publication Date(Web):
DOI:10.1002/macp.201100574
No abstract is available for this article.
Co-reporter:Dominik Voll;Thomas Junkers
Journal of Polymer Science Part A: Polymer Chemistry 2012 Volume 50( Issue 14) pp:2739-2757
Publication Date(Web):
DOI:10.1002/pola.26076
Abstract
The current article contains a review of the electrospray ionization-mass spectrometry characterization of polymers prepared via thermal- and photoinitiation processes. The used analysis method permits direct access to detailed endgroup information. For a qualitative and quantitative endgroup analysis, sophisticated methods have been developed which provide a detailed image of the incorporation propensity of thermally as well as photolytically generated radicals at the polymer chain termini. Such a post-mortem analysis of polymeric materials specifically allows for the quantification of the ability of radical fragments to initiate polymerization processes. Herein, the most recent progress in the field of mass spectrometric radical reactivity mapping is outlined and open questions as well as future directions are discussed. © 2012 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem, 2012
Co-reporter:Anna-Marie Zorn
Journal of Polymer Science Part A: Polymer Chemistry 2012 Volume 50( Issue 12) pp:2366-2377
Publication Date(Web):
DOI:10.1002/pola.26011
Abstract
In the present study, n-butyl acrylate macromonomer (BAMM) (Mn = 1900 g mol−1; PDI = 1.96) has been synthesized via a high-temperature polymerization process. Subsequently, the olefinic termini of the BAMM have been transformed into a diol via a dihydroxylation process using KMnO4 as an oxidizing agent. The OH-terminated macroinitiator pBA(OH)2 has subsequently been employed for the ring-opening polymerization (ROP) of ε-caprolactone via various catalytic systems, that is, organo-(1,5,7-triazabicyclo[4.4.0]dec-5-ene), metal (tin(II) 2-ethylhexanoate), and enzymatic catalysis (Novozym® 435). The obtained pBA-b-pCL block copolymers and the initiation efficiency of the BAMM macroinitiator have been investigated via size exclusion chromatography (SEC), electrospray ionization–mass spectrometry (ESI-MS) hyphenated with SEC and liquid chromatography at the critical conditions of both poly(ε-caprolactone) (pCL) and pBA. The in vitro enzyme catalysis (eROP) approach proved to be the most efficient catalysis system due to minor transesterification side reactions during the polymerization process. However, side reactions such as transesterifications occur in each catalytic system and—while they cannot be suppressed—they can be minimized. The species generated during the eROP process include the desired block copolymer pBA-b-pCL as main species as well as pCL homopolymer and residual macroinitiator pBA(OH)2. © 2012 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem, 2012
Co-reporter:Christoph Herfurth;Dominik Voll;Jens Buller;Jan Weiss;André Laschewsky
Journal of Polymer Science Part A: Polymer Chemistry 2012 Volume 50( Issue 1) pp:108-118
Publication Date(Web):
DOI:10.1002/pola.24994
Abstract
We report on the controlled free radical homopolymerization of 1-ferrocenylethyl acrylate as well as of three new ferrocene bearing monomers, namely 4-ferrocenylbutyl acrylate, 2-ferrocenylamido-2-methylpropyl acrylate, and 4-ferrocenylbutyl methacrylate, by the RAFT technique. For comparison, the latter monomer was polymerized using ATRP, too. The ferrocene containing monomers were found to be less reactive than their analogues free of ferrocene. The reasons for the low polymerizability are not entirely clear. As the addition of free ferrocene to the reaction mixture did not notably affect the polymerizations, sterical hindrance by the bulky ferrocene moiety fixed on the monomers seems to be the most probable explanation. Molar masses found for 1-ferrocenylethyl acrylate did not exceed 10,000 g mol−1, while for 4-ferrocenylbutyl (meth)acrylate molar masses of 15,000 g mol−1 could be obtained. With PDIs as low as 1.3 in RAFT polymerization of the monomers, good control over the polymerization was achieved. © 2011 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem, 2012
Co-reporter:Christoph J. Dürr;Sebastian G. J. Emmerling;Andreas Kaiser;Sven Brau;Axel K. T. Habicht;Michael Klimpel
Journal of Polymer Science Part A: Polymer Chemistry 2012 Volume 50( Issue 1) pp:174-180
Publication Date(Web):
DOI:10.1002/pola.25033
Abstract
The synthesis of acrylonitrile-butadiene rubbers (NBRs) via trithiocarbonate-mediated reversible addition fragmentation chain transfer (RAFT) polymerization of acrylonitrile (ACN) and 1,3-butadiene (BD) in solution under azeotropic conditions (38/62) was investigated for a broad range of common solvents: N,N-dimethylacetamide (DMAc), chlorobenzene, 1,4-dioxane, tert-butanol, isobutyronitrile, toluene, trimethylacetonitrile, dimethyl carbonate, acetonitrile, methyl acetate, acetone, and tert-butyl methyl ether. The gravimetrically determined conversions for the free radical polymerizations of ACN/BD after 22 h at 100 °C were in the range of 15% for methyl acetate to 35% for DMAc. The origin of the differences in conversion is attributed to the unequal decomposition behavior of the employed azo initiator 2,2′-azobis(N-butyl-2-methylpropionamide) (1) in the solvents under investigation, as determined by ultraviolet–visible (UV–vis) spectroscopy. Relative decomposition of 1 in solution (0.1 mol L−1) at 100 °C was calculated from the UV–vis spectra for selected solvents. 90% of 1 in DMAc was decomposed after 22 h, 83% in tert-butanol, 57% in 1,4-dioxane, 53% in isobutyronitrile, 45% in chlorobenzene, and 21% in toluene. The evolution of molecular weight with conversion using the initiator 1 was in accordance with the theoretically expected values, regardless of the solvent studied. Moreover, the RAFT-mediated copolymerization of ACN/BD in DMAc with azo initiators 1, 1-[(1-cyano-1-methylethyl)azo]formamide (2) and 1,1′-azobis(cyclohexanecarbonitrile) (3) was investigated. A strong deviation from the linear evolution of molecular weight due to a fast decomposition of these initiators – congruent with high primary radical delivery rates – at the selected temperature was observed when using 2 and 3. The deviation was not observed when using 1. © 2011 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem, 2012
Co-reporter:Guillaume Delaittre, Mathias Dietrich, James P. Blinco, Astrid Hirschbiel, Michael Bruns, Leonie Barner, and Christopher Barner-Kowollik
Biomacromolecules 2012 Volume 13(Issue 5) pp:
Publication Date(Web):March 16, 2012
DOI:10.1021/bm3001364
Co-reporter:Alexer H. Soeriyadi;Vanessa Trouillet;Francesca Bennet;Michael Bruns;Michael R. Whittaker;Cyrille Boyer;Philip J. Barker;Thomas P. Davis
Journal of Polymer Science Part A: Polymer Chemistry 2012 Volume 50( Issue 9) pp:1801-1811
Publication Date(Web):
DOI:10.1002/pola.25947
Abstract
The present study investigates the degradation behavior of various high-molecular-weight acrylic polymers (50,000 < Mn/g mol−1 < 100,000), namely poly(methyl methacrylate) (PMMA), poly(n-butyl methacrylate) (PBMA), poly(n-butyl acrylate) (PBA), and poly(lauryl methacrylate) (PLMA), under extreme environmental conditions. These polymers were synthesized via various polymerization techniques to create different end-groups. The polymers chosen are readily applicable in the formulation of surface coatings and were degraded under conditions which replicate the harsh Australian climate, where surface coatings may reach temperatures of up to 95 °C and are exposed to broad-spectrum UV radiation of up to 1 kW m−2. The degradation behavior of the polymeric materials on their surface was followed via ATR-IR spectroscopy, high resolution FTIR microscopy, and X-ray photoelectron spectroscopy. The extent of the observed thermal and photo-oxidation is directly related to the length of the ester side group, with the degradation susceptibility decreasing in the order of PLMA > PBMA/PBA > PMMA, with PMMA still stable even after 5 months exposure to the harshest condition used (UV light at 95 °C). The general degradation mechanism involves the loss of the ester side groups to form methacrylic acid followed by cross-linking. The effect of the variable end groups was found to be minimal. The results from this study are in good agreement with previous studies of low-molecular-weight model polymers under identical conditions. © 2012 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem, 2012
Co-reporter:Christina Schmid, Jana Falkenhagen, Timo F. Beskers, Le-Thu T. Nguyen, Manfred Wilhelm, Filip E. Du Prez, and Christopher Barner-Kowollik
Macromolecules 2012 Volume 45(Issue 16) pp:6353-6362
Publication Date(Web):August 6, 2012
DOI:10.1021/ma301117k
The detailed characterization of poly(styrene)-b-poly(tetrahydrofuran) (pS-b-pTHF) multiblock copolymers (17800 g mol–1 ≤ Mn ≤ 46800 g mol–1) generated via urethane linkages is presented. The synthesis of the block copolymers is enabled via a mechanistic switch of the thiocarbonyl thio end group of a poly(styrene) to dihydroxyl terminated polymers that subsequently react with a diisocyanate terminated polytetrahydrofuran based prepolymer to form multiblock copolymer structures. The characterization of the multiblock copolymers and their substructures includes size exclusion chromatography (SEC), liquid chromatography at critical conditions (LCCC), nuclear magnetic resonance (NMR), and infrared (IR) spectroscopy as well as matrix-assisted laser desorption/ionization (MALDI) mass spectrometry. To obtain even further details of the polymer size and its composition, SEC with triple detection as well as newly developed SEC coupled online to IR spectroscopy was carried out. The quantification of the average block fractions via online SEC-IR (41–61 mol % pTHF) is in very good agreement with the results obtained via NMR spectroscopy (39–66 mol % pTHF).
Co-reporter:Thomas Pauloehrl;Dr. Guillaume Delaittre;Volker Winkler;Dr. Alexer Welle;Dr. Michael Bruns;Dr. Hans G. Börner;Dr. Alexra M. Greiner;Dr. Martin Bastmeyer;Dr. Christopher Barner-Kowollik
Angewandte Chemie International Edition 2012 Volume 51( Issue 4) pp:1071-1074
Publication Date(Web):
DOI:10.1002/anie.201107095
Co-reporter:Thomas J. A. Wolf, Dominik Voll, Christopher Barner-Kowollik, and Andreas-Neil Unterreiner
Macromolecules 2012 Volume 45(Issue 5) pp:2257-2266
Publication Date(Web):February 28, 2012
DOI:10.1021/ma202673q
The excited states and dynamics of the three triplet radical photoinitiators benzoin (2-hydroxy-1,2-diphenylethanone, Bz), 2,4,6-trimethylbenzoin (2-hydroxy-1-mesityl-2-phenylethanone, TMB), and mesitil (1,2-bis(2,4,6-trimethylphenyl)-1,2-ethanedione, Me)—employed in our previous studies for quantifying net initiation efficiencies in pulsed laser polymerization with methacrylate monomers [Voll, D.; Junkers, T.; Barner-Kowollik, C. Macromolecules2011, 44, 2542–2551]—are investigated via both femtosecond transient absorption (TA) spectroscopy and density functional theory (DFT) methods to elucidate the underlying mechanisms causing different initiation efficiencies when excited at 351 nm. Bz and TMB are found to have very similar properties in the calculated excited states as well as in the experimentally observed dynamics. After excitation into the first excited singlet state (S1) Bz and TMB undergo rapid intersystem crossing (ISC). The ISC can compete with ultrafast internal conversion (IC) processes because an excited triplet state (Tn) of nearly the same energy is present in both cases. ISC is therefore the dominating depopulation channel of S1, and subsequent α-cleavage to produce radicals takes place on the picosecond time scale. In contrast, Me is excited into the second excited singlet state (S2). In this case no isoenergetic triplet state is available, which inhibits ISC from competing with ultrafast deactivation processes. ISC is therefore assigned to be a minor deactivation channel in Me. Employing these findings, quantitative photoinitiation efficiency relations of Bz, TMB, and Me obtained by pulsed laser polymerization can be directly correlated with the relative TA intensities found in the femtosecond experiments. The ISC efficiency is thus a critical parameter for evaluating the overall photoinitiation efficiency and demonstrates that the employment of the herein presented method represents a powerful tool for attaining a quantitative picture on the suitability of a photoinitiator.
Co-reporter:Thomas Pauloehrl;Dr. Guillaume Delaittre;Dr. Michael Bruns;Maria Meißler;Dr. Hans G. Börner;Dr. Martin Bastmeyer;Dr. Christopher Barner-Kowollik
Angewandte Chemie International Edition 2012 Volume 51( Issue 36) pp:9181-9184
Publication Date(Web):
DOI:10.1002/anie.201202684
Co-reporter:Thomas Pauloehrl;Dr. Guillaume Delaittre;Volker Winkler;Dr. Alexer Welle;Dr. Michael Bruns;Dr. Hans G. Börner;Dr. Alexra M. Greiner;Dr. Martin Bastmeyer;Dr. Christopher Barner-Kowollik
Angewandte Chemie 2012 Volume 124( Issue 4) pp:1096-1099
Publication Date(Web):
DOI:10.1002/ange.201107095
Co-reporter:Thomas Pauloehrl;Dr. Guillaume Delaittre;Dr. Michael Bruns;Maria Meißler;Dr. Hans G. Börner;Dr. Martin Bastmeyer;Dr. Christopher Barner-Kowollik
Angewandte Chemie 2012 Volume 124( Issue 36) pp:9316-9319
Publication Date(Web):
DOI:10.1002/ange.201202684
Co-reporter:Matthias Winkler, Jan O. Mueller, Kim K. Oehlenschlaeger, Lucas Montero de Espinosa, Michael A. R. Meier, and Christopher Barner-Kowollik
Macromolecules 2012 Volume 45(Issue 12) pp:5012-5019
Publication Date(Web):June 7, 2012
DOI:10.1021/ma3007043
Within the current contribution, we introduce two strategies for the catalyst-free, modular, ambient temperature synthesis of ABC triblock copolymers via photoinduced Diels–Alder reactions. On the one hand, the 2-formyl-3-methylphenoxy (FMP) moiety (a second generation photoenol precursor) was employed for orthogonal polymer–polymer conjugations using terminal acrylates of diblock copolymers synthesized via acyclic-diene-metathesis (ADMET) polymerizations to directly prepare triblock copolymers. On the other hand, the disparate reactivity of 2,5-dimethylbenzophenone (first generation photoenol) and the FMP moiety was exploited to selectively synthesize complex triblock copolymers (6.5 kDa≤ Mn ≤11.5 kDa, 1.16≤ PDI ≤ 1.30) via a sequential one pot approach utilizing the extraordinary orthogonality of the photoinduced Diels–Alder reaction. Polymers functionalized with a photoenol (second generation) moiety were employed for conjugation reactions with polymers featuring an acrylate terminus, while polymers having a photoenol (first generation) end group were employed for selective conjugations of maleimide functional polymers. In this context, the selective head-to-tail ADMET polymerization was employed as a straightforward methodology for the preparation of bifunctional polymers having a terminal acrylate and a photoenol end-group.
Co-reporter:Christina Schmid, Steffen Weidner, Jana Falkenhagen, and Christopher Barner-Kowollik
Macromolecules 2012 Volume 45(Issue 1) pp:87-99
Publication Date(Web):December 20, 2011
DOI:10.1021/ma2022452
A recently introduced procedure involving a mechanistic switch from reversible addition–fragmentation chain transfer (RAFT) polymerization to ring-opening polymerization (ROP) to form diblock copolymers is applied to synthesize ABA (star) block copolymers. The synthetic steps include the polymerization of styrene with R-group designed RAFT agents, the transformation of the thiocarbonyl thio end groups into OH functionalities, and their subsequent chain extension by ROP. The obtained linear ABA poly(ε-caprolactone)-block-poly(styrene)-block-poly(ε-caprolactone) (pCL-b-pS-b-pCL) (12 500 g mol–1 ≤ Mn ≤ 33 000 g mol–1) and the star-shaped poly(styrene)-block-poly(ε-caprolactone) (Mn = 36 000 g mol–1) copolymers were analyzed by size exclusion chromatography (SEC), nuclear magnetic resonance (NMR), infrared (IR) spectroscopy, and matrix-assisted laser desorption/ionization (MALDI) mass spectrometry. The focus of the current study is on the detailed characterization of the ABA (star) block polymers via multidimensional chromatographic techniques specifically high performance liquid chromatography coupled to size exclusion chromatography (HPLC-SEC). In particular, we demonstrate the first time separation of poly(ε-caprolactone) (pCL) homopolymer and additionally poly(styrene) (pS) from the ABA poly(ε-caprolactone)-b-poly(styrene)-b-poly(ε-caprolactone) and star-shaped poly(styrene)-b-poly(ε-caprolactone) block copolymer utilizing critical conditions (CC) for pCL with concomitant gradient elution liquid chromatography (GELC).
Co-reporter:Guillaume Delaittre, Thomas Pauloehrl, Martin Bastmeyer, and Christopher Barner-Kowollik
Macromolecules 2012 Volume 45(Issue 4) pp:1792-1802
Publication Date(Web):February 8, 2012
DOI:10.1021/ma202670d
A new set of monomers is presented in order to incorporate thiols into radical polymers using a protecting chemistry/photocleavage route. The (co)polymerization kinetics of an o-nitrobenzyl thioether-containing acrylamide derivative are reported. The presence of the o-nitrobenzyl moiety is found to strongly affect the polymerization. Nevertheless, water-soluble copolymers with N,N-dimethylacrylamide (DMAAm) as a comonomer are obtained either by free radical polymerization (10 000 ≤ Mn ≤ 17 500 g mol–1; 1.5 ≤ PDI ≤ 1.8) or by reversible addition–fragmentation transfer (RAFT)-mediated controlled/living radical polymerization (2000 ≤ Mn ≤ 5700 g mol–1; 1.1 ≤ PDI ≤ 1.2). Deprotection under UV light (λ = 366 nm) at ambient temperature is followed by UV/vis monitoring of the protecting group release, which proceeds to completion between 40 min and 2 h within the studied range of concentration as demonstrated by 1H NMR spectroscopy. Thiol–maleimide addition is subsequently carried out and found to proceed with a nearly quantitative yield (ca. 90%) as measured by 1H NMR. Different block copolymers (9400 ≤ Mn ≤ 16 500 g mol–1; 1.3 ≤ PDI ≤ 1.4) with a PDMAAm water-soluble block, a polystyrene hydrophobic block, or a poly(N-isopropylacrylamide) thermosensitive block as the first segment and possessing the photoreleasable thiol moieties in the second block are subsequently synthesized by RAFT-mediated polymerization. We finally demonstrate the orthogonal sequential deprotection and reaction with benzyl maleimide of two different thiol species originating from the thiocarbonylthio functionality and the o-nitrobenzyl protected lateral groups, respectively.
Co-reporter:James P. Blinco;Vanessa Trouillet;Michael Bruns;Peter Gerstel;Hartmut Gliemann
Advanced Materials 2011 Volume 23( Issue 38) pp:4435-4439
Publication Date(Web):
DOI:10.1002/adma.201102875
Co-reporter:Mathias Glassner, Kristian Kempe, Ulrich S. Schubert, Richard Hoogenboom and Christopher Barner-Kowollik
Chemical Communications 2011 vol. 47(Issue 38) pp:10620-10622
Publication Date(Web):01 Sep 2011
DOI:10.1039/C1CC14075B
An efficient method for the preparation of cyclopentadienyl endcapped poly(2-ethyl-2-oxazoline) (PEtOx–Cp) via cationic ring-opening polymerization utilizing sodium cyclopentadienide as a termination agent is presented. Subsequent Diels–Alder reactions with N-substituted maleimides proceed quantitatively at ambient temperature. A block copolymer (PEtOx-b-PEG) is prepared employing maleimide terminated poly(ethylene glycol).
Co-reporter:Edgar H. H. Wong, Ozcan Altintas, Martina H. Stenzel, Christopher Barner-Kowollik and Thomas Junkers
Chemical Communications 2011 vol. 47(Issue 19) pp:5491-5493
Publication Date(Web):11 Apr 2011
DOI:10.1039/C1CC10322A
A synthetic strategy employing nitrones as radical spin traps is presented on the example of the efficient generation of novel dendrimersvia a combination of radical and classical ‘click’ chemistry.
Co-reporter:Anna-Marie Zorn, Michael Malkoch, Anna Carlmark and Christopher Barner-Kowollik
Polymer Chemistry 2011 vol. 2(Issue 5) pp:1163-1173
Publication Date(Web):14 Mar 2011
DOI:10.1039/C0PY00411A
The combination of dendrons and high temperature acrylate polymerization represents a viable route to form dendronized macromonomers. Dendronized acrylates based on 2,2-bis(hydroxymethyl)propionic acid (bis-MPA) were synthesized using dendrimer synthesis and click chemistry (copper catalyzed azide alkyne cycloaddition (CuAAC)). The synthesis was carried out up to the 3rd generation and with a carbon spacer length of 6 or 9 between the acrylic function and the dendron core. These dendronized acrylates were subjected to auto-initiated high temperature acrylate polymerization. The polymerization was performed at 140 °C in a 5 wt% solution of hexyl acetate with a 2,2′-azobis(isobutyronitrile) (AIBN) concentration of 5 × 10−3 g mol−1. The vinyl terminated polymers were in-depth characterized viasize exclusion chromatography (SEC) and size exclusion chromatography coupled to electrospray ionization mass spectrometry (SEC-ESI-MS) to assess the generated product spectrum and the efficiency of the process. The achievable number average molecular weight, Mn, was between 1700 and 4400 g mol−1. The degree of polymerization, DPn, decreases with increasing generations of the dendronized acrylates from 6.3 to 3.4. The purity of vinyl terminated oligomers containing a geminal double bond is up to 83%, with the dendronized acrylates of the 1st generation providing the best result. Moderate deprotection of the acetonide groups occurred spontaneously during the macromonomer formation process and reached its maximum at generation 3.
Co-reporter:Ozcan Altintas, Umit Tunca and Christopher Barner-Kowollik
Polymer Chemistry 2011 vol. 2(Issue 5) pp:1146-1155
Publication Date(Web):16 Feb 2011
DOI:10.1039/C0PY00395F
Hamilton wedge (HW) end-functionalized poly(styrene) (PS–HW, Mn = 5400 g mol−1, PDI = 1.06), HW mid-chain functionalized poly(styrene) (PS–HW–PS, Mn = 4600 g mol−1, PDI = 1.04), cyanuric acid (CA) end-functionalized poly(styrene) (PS–CA, Mn = 3700 g mol−1, PDI = 1.04) and CA end-functionalized poly(methyl methacrylate) (PMMA–CA, Mn = 8500 g mol−1, PDI = 1.13) precursors were successfully synthesized via a combination of atom transfer radical polymerization (ATRP) and copper catalyzed azide–alkyne cycloaddition (CuAAC). The precursor polymers were characterized viasize exclusion chromatography (SEC) and 1H NMR with respect to both molecular weight and structure. Supramolecular homopolymer (PS–HW·PS–CA), block copolymer (PS–HW·PMMA–CA), star polymer (PS–HW–PS·PS–CA) as well as miktoarm star polymer (PS–HW–PS·PMMA–CA) were formed in solution in high yields at ambient temperature (association close to 89% for PS–HW·PS–CA, 90% for PS–HW–PS·PS–CA and 98% for PS–HW–PS·PMMA–CA) via H-bonding between the orthogonal recognition units, HW and CA. The formation of supramolecular polymers was confirmed via1H NMR at ambient temperature in deuterated methylene chloride (CD2Cl2) solution.
Co-reporter:Edgar H. H. Wong, Thomas Junkers and Christopher Barner-Kowollik
Polymer Chemistry 2011 vol. 2(Issue 5) pp:1008-1017
Publication Date(Web):24 Jan 2011
DOI:10.1039/C0PY00377H
Nitrones are arguably one of the most efficient compounds with multi-functional capabilities, acting as both radical spin traps and 1,3-dipoles. In contrast to their potential synthetic versatility, the application of nitrones in synthetic polymer chemistry is still in its infancy and its true potential has yet to be fully explored. The current report thus serves as a highlight and provides an update on the current status of nitrones in macromolecular synthesis.
Co-reporter:Andrew J. Inglis and Christopher Barner-Kowollik
Polymer Chemistry 2011 vol. 2(Issue 1) pp:126-136
Publication Date(Web):23 Aug 2010
DOI:10.1039/C0PY00189A
The quantification of the efficient ultra-rapid modular synthesis of block copolymersviatwo-dimensional chromatography and a comparably accurate, low-cost deconvolution of size exclusion chromatography traces is reported. Cyclopentadienyl-capped poly(methyl methacrylate) (PMMA, 97% functionality) and a series of pyridin-2-yldithioformate-capped poly(isobornyl acrylate)s (PiBoA, 93–95% functionality) were synthesiszed via atom transfer radical polymerization (ATRP) and reversible addition fragmentation chain transfer (RAFT) polymerization respectively. The corresponding block copolymers were generated by simply stirring a chloroform solution of PMMA, PiBoA and acidic catalyst for 10 min at ambient temperature. The crude block copolymer mixtures were directly analyzed by size exclusion chromatography (SEC), liquid adsorption chromatography at critical conditions (LACCC) and 2D LACCC-SEC to visualize the efficiency of the ultra rapid conjugation reaction. The quantitative analysis via the 2D LACCC-SEC yielded the composition of the block copolymer mixtures which were in excellent agreement with predicted values, thus indicating quantitative conjugation efficiency. In all cases, the crude block copolymer mixtures contained at least 94 wt% block copolymer. Furthermore, a low-cost deconvolution method that may be applied to conventional SEC traces was found to provide comparable composition data.
Co-reporter:Mathias Glassner, James P. Blinco and Christopher Barner-Kowollik
Polymer Chemistry 2011 vol. 2(Issue 1) pp:83-87
Publication Date(Web):04 Oct 2010
DOI:10.1039/C0PY00267D
Poly(styrene)-block-poly(ethylene oxide) copolymers synthesized via the combination of reversible addition fragmentation chain transfer (RAFT) polymerization and hetero Diels–Alder (HDA) cycloaddition can be cleaved in the solid state by a retro-HDA reaction occurring at 90 °C. Nanoporous films can be prepared from these polymers using a simple heating and washing procedure.
Co-reporter:Peter Gerstel
Macromolecular Rapid Communications 2011 Volume 32( Issue 5) pp:444-450
Publication Date(Web):
DOI:10.1002/marc.201000730
Co-reporter:Mathias Glassner;James P. Blinco
Macromolecular Rapid Communications 2011 Volume 32( Issue 9-10) pp:724-728
Publication Date(Web):
DOI:10.1002/marc.201100094
Co-reporter:Till Gruendling;Kim K. Oehlenschlaeger;Elena Frick;Mathias Glassner;Christina Schmid
Macromolecular Rapid Communications 2011 Volume 32( Issue 11) pp:807-812
Publication Date(Web):
DOI:10.1002/marc.201100159
Co-reporter:Edgar Espinosa;Mathias Glassner;Christophe Boisson;Franck D'Agosto
Macromolecular Rapid Communications 2011 Volume 32( Issue 18) pp:1447-1453
Publication Date(Web):
DOI:10.1002/marc.201100310
Co-reporter:Thomas Junkers;Michelle L. Coote
Macromolecular Rapid Communications 2011 Volume 32( Issue 23) pp:1891-1898
Publication Date(Web):
DOI:10.1002/marc.201100494
Abstract
In a recent article (W. Meiser, M. Buback, Assessing the RAFT Equilibrium Constant via Model Systems: An EPR Study, Macromol. Rapid Commun.2011, 18, 1490-1494), it is claimed that evidence is found that unequivocally proves that quantum mechanical calculations assessing the equilibrium constant and fragmentation rate coefficients in dithiobenzoate-mediated reversible addition fragmentation transfer (RAFT) systems are beset with a considerable uncertainty. In the present work, we show that these claims made by Meiser and Buback are beset with a model dependency, as a critical key parameter in their data analysis – the addition rate coefficient of the radicals attacking the C=S double bond in the dithiobenzoate – induces a model insensitivity into the data analysis. Contrary to the claims made by Meiser and Buback, their experimental results can be brought into agreement with the quantum chemical calculations if a lower addition rate coefficient of cyanoisopropyl radicals (CIP) to the CIP dithiobenzoate (CPDB) is assumed. To resolve the model dependency, the addition rate coefficient of CIP radicals to CPDB needs to be determined as a matter of priority.
Co-reporter:James P. Blinco, Andreas Greiner, Christopher Barner-Kowollik, Seema Agarwal
European Polymer Journal 2011 Volume 47(Issue 1) pp:111-114
Publication Date(Web):January 2011
DOI:10.1016/j.eurpolymj.2010.10.025
In this communication we provide the most recent results on RAFT-mediated ring-closing polymerization of diallyldimethylammonium chloride (DADMAC). The polymerization was carried out in aqueous solution employing 2,2′-azobis(2-methylpropionamidine)-dihydrochloride as the free radical initiator and trithiocarbonate RAFT agent (2-{[(dodecylsulfanyl)carbonothioyl sulfanyl]}propanoic acid, DoPAT) as the controlling RAFT agent. The results show that – while the system is not as completely controlled as previously described – it is nevertheless possible to mediate the polymerization of DADMAC and impart some living characteristics onto the system. The initial study on the RAFT-mediated polymerization of DADMAC may have overestimated the degree of livingness within this reaction. However, it is possible – at low conversions – for some living characteristics to be observed, as the evolution of molecular weight with conversion is linear. In addition, polymers with a reasonably narrow polydispersity can be isolated.
Co-reporter:Corinna M. Preuss
Macromolecular Theory and Simulations 2011 Volume 20( Issue 8) pp:700-708
Publication Date(Web):
DOI:10.1002/mats.201100033
Co-reporter:Christiane Lang;Dominik Voll;Andrew J. Inglis;Nico Dingenouts;Anja S. Goldmann;Leonie Barner
Macromolecular Chemistry and Physics 2011 Volume 212( Issue 8) pp:831-839
Publication Date(Web):
DOI:10.1002/macp.201100010
Co-reporter:Boris Bitsch;Shiping Zhu
Macromolecular Reaction Engineering 2011 Volume 5( Issue 1) pp:55-68
Publication Date(Web):
DOI:10.1002/mren.201000034
Co-reporter:Till Gruendling, Michael Kaupp, James P. Blinco, and Christopher Barner-Kowollik
Macromolecules 2011 Volume 44(Issue 1) pp:166-174
Publication Date(Web):November 8, 2010
DOI:10.1021/ma101893u
We report the photoinduced conjugation of polymers synthesized via reversible addition−fragmentation chain transfer (RAFT) polymerization with a number of low molecular weight (functional) olefins. Upon irradiation of a solution of an aliphatic alkene and the benzyl dithioacetic acid ester (CPDA) or dodecyl trithiocarbonate (DoPAT) functional poly(alkyl acrylate) at the absorption wavelength of the thiocarbonyl group (315 nm), incorporation of the alkene at the polymer chain-end occurred. The most efficient systems identified with regard to the rate of reaction and yield were poly(butyl acrylate)/CPDA/ethyl vinyl ether (78% monoinsertion product after 1 h) and poly(butyl acrylate)/CPDA/1-pentene (73% insertion product after 7 h) at ambient temperature. An in-depth analysis of the reaction mechanism by 1H NMR and online size-exclusion chromatography-electrospray ionization tandem mass spectrometry (SEC/ESI−MSn) revealed that a possible [2 + 2] photoaddition mechanism of conjugation does not take place. Instead, fast β-cleavage of the photoexcited RAFT-end group with subsequent radical addition of an alkene was observed for all employed systems. The presented reaction thus provides a means of spatial and temporal control for the conjugation of alkenes to thiocarbonyl thio-capped macromolecules via the use of UV radiation.
Co-reporter:Thomas Junkers;Lin Zang;Edgar H.H. Wong;Nico Dingenouts
Journal of Polymer Science Part A: Polymer Chemistry 2011 Volume 49( Issue 22) pp:4841-4850
Publication Date(Web):
DOI:10.1002/pola.24970
Abstract
The preparation of ABA-type block copolymers via tandem enhanced spin capturing polymerization (ESCP) and nitroxide-mediated polymerization (NMP) processes is explored in-depth. Midchain alkoxyamine functional polystyrenes (Mn = 6200, 12,500 and 19,900 g mol−1) were chain extended with styrene as well as tert-butyl acrylate at elevated temperature NMP conditions (T = 110 °C) generating a tandem ESCP-NMP sequence. Although the chain extensions and thus the block copolymer formation processes function well (yielding in the case of the chain extension with styrene number average molecular weights of up to 20,800 g mol−1 (PDI = 1.22) when the 6200 g mol−1 precursor is used and up to 67,500 g mol−1 (PDI = 1.36) when the 19,900 g mol−1 precursor is used and 21,600 g mol−1 (PDI = 1.17) as well as 37,100 g mol−1 (PDI = 1.21) for the tert-butyl acrylate chain extensions for the 6200 and 12,500 g mol−1 precursors, respectively), it is also evident that the efficiency of the block copolymer formation process decreases with an increasing chain length of the ESCP precursor macromolecules (i.e., for the 19,900 g mol−1 ESCP precursor no efficient chain extension with tert-butyl acrylate can be observed). For the polystyrene-block-tert-butyl acrylate-block-polystyrene polymers, the molecular weights were determined via triple detection SEC using light scattering and small-angle X-ray scattering. © 2011 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem, 2011.
Co-reporter:Ozcan Altintas;Tobias Rudolph
Journal of Polymer Science Part A: Polymer Chemistry 2011 Volume 49( Issue 12) pp:2566-2576
Publication Date(Web):
DOI:10.1002/pola.24688
Abstract
Well-defined heterotelechelic poly(styrene) carrying thymine/diaminopyridine (DAP) (Mn,SEC = 9300, PDI = 1.04) and Hamilton wedge (HW)/cyanuric acid (CA) (Mn,SEC = 8200, PDI = 1.04) bonding motifs are prepared via a combination of controlled/living radical polymerization and copper catalyzed azide/alkyne “click” chemistry and are subsequently self-assembled as single chains to emulate—on a simple level—the self-folding behavior of natural biomacromolecules. Hydrogen nuclear magnetic resonance (1H NMR) in deuterated dichloromethane and dynamic light scattering analyses provides evidence for the hydrogen bonding interactions between the α-thymine and ω-DAP as well as α-CA and ω-HW chain ends of the heterotelechelic polymers leading to circular entropy driven single chain self-assembly. This study demonstrates that the choice of NMR solvent is important for obtaining well-resolved NMR spectra of the self-assembled structures. In addition, steric effects on the HW can affect the efficiency of the self-assembly process. © 2011 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem, 2011
Co-reporter:Christopher Barner-Kowollik;Thomas Junkers
Journal of Polymer Science Part A: Polymer Chemistry 2011 Volume 49( Issue 5) pp:1293-1297
Publication Date(Web):
DOI:10.1002/pola.24546
Co-reporter:Alexer H. Soeriyadi;Francesca Bennet;Michael R. Whittaker;Philip J. Barker;Thomas P. Davis
Journal of Polymer Science Part A: Polymer Chemistry 2011 Volume 49( Issue 4) pp:848-861
Publication Date(Web):
DOI:10.1002/pola.24492
Abstract
This study investigates the degradation behavior of poly(n-butyl methacrylate) (p(nBMA)), poly(tert-butyl methacrylate) (p(tBMA)), and poly(hexafluoro butyl methacrylate) (p(HFBMA)) on a molecular level under extreme environmental conditions. The polymers chosen are readily applicable in the formulation of surface coatings and were degraded under conditions which replicated the harsh Australian climate, in which surface coatings may reach temperatures of up to 95 °C and are exposed to broad-spectrum UV radiation of up to 1 kW m−2. The degradation profiles were mapped with high-resolution electrospray ionization mass spectrometry (ESI-MS) with a LCQ quadrupole ion trap mass analyzer, with the peak assignments confirmed to within 3 ppm using ESI-MS with a LTQ-Orbitrap mass detector. It was found that in all the butyl ester polymers analyzed herein—regardless of their tertiary side-chain structure—the loss of the butyl ester group and subsequent formation of acid side groups are a component of the overall degradation pathway of poly(butyl methacrylate)s under these harsh conditions. However, it is also demonstrated that the magnitude of this pathway is intimately linked to the side-chain structure with the propensity for degradation decreasing in the order p(tBMA) > p(nBMA) > p(HFBMA). The degradation mechanisms identified in this study, in combination with the previous end-group degradation studies of poly(methyl methacrylate) and poly(n-butyl acrylate), have allowed a much deeper understanding of the molecular degradation behavior of poly(acrylate)s and poly(methacrylate)s in an extreme natural environment. © 2010 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem, 2011
Co-reporter:Nicolas Zydziak, Christof Hübner, Michael Bruns, and Christopher Barner-Kowollik
Macromolecules 2011 Volume 44(Issue 9) pp:3374-3380
Publication Date(Web):April 7, 2011
DOI:10.1021/ma200107z
Without previous modification, single-walled carbon nanotubes (SWCNTs) react as dienophiles in a single-step Diels−Alder [4 + 2] cycloaddition with diene terminal polymer strands. Cyclopentadienyl-capped poly(methyl methacrylate) (Mn = 2900 g mol−1, PDI = 1.2) was grafted onto SWCNTs under mild conditions at ambient temperature as well as at 80 °C in the absence of any catalyst. Thermogravimetric analysis, elemental analysis, and X-ray photoelectron spectroscopy confirm the success of the reaction and allow to estimate the grafting density of the polymer chains on the SWCNTs via the above three independent methods (average grafting density of 0.064 mmol g−1 (0.029 chains nm−2) for samples reacted at ambient temperature and 0.086 mmol g−1 (0.039 chains nm−2) for samples reacted at 80 °C). In addition, high-resolution transmission electron microscopy images confirmed the presence of an amorphous polymer layer (∼3 nm) around the SWCNTs after functionalization.
Co-reporter:Bernhard V. K. J. Schmidt, Martin Hetzer, Helmut Ritter, and Christopher Barner-Kowollik
Macromolecules 2011 Volume 44(Issue 18) pp:7220-7232
Publication Date(Web):August 25, 2011
DOI:10.1021/ma2011969
The first aqueous reversible addition–fragmentation transfer (RAFT) polymerization of N,N-dimethylacrylamide (DMAAm), N,N-diethylacrylamide (DEAAm), and N-isopropylacrylamide (NIPAAm) utilizing host/guest complexes of cyclodextrin and hydrophobic chain transfer agents (CTAs) at 25 °C is described. Three novel guest-functionalized CTAs, namely 4-(tert-butyl)phenyl 2-(((ethylthio)carbonothioyl)thio)-2-methylpropanoate, bis(4-tert-butyl)benzyl carbonotrithioate, and benzyl (3-((4-(tert-butyl)phenyl)amino)-3-oxopropyl)carbonotrithioate, were synthesized and employed in aqueous RAFT polymerizations. The presented technique allows for the facile preparation of hydrophilic polymers with hydrophobic end groups in aqueous environments. The living/controlled radical polymerization afforded high molecular masses (7500 ≤ Mn ≤ 116 000 g mol–1 for poly(DMAAm), 2500 ≤ Mn ≤ 150 000 g mol–1 for poly(DEAAm), and 4000 ≤ Mn ≤ 50 000 g mol–1 for poly(NIPAAm)) with low PDIs (1.06 ≤ PDI ≤ 1.54 for poly(DMAAm), 1.05 ≤ PDI ≤ 1.39 for poly(DEAAm), and 1.15 ≤ PDI ≤ 1.46 for poly(NIPAAm)). To confirm the living character of the polymerizations, kinetic measurements were undertaken that evidence a linear evolution of molecular weight with conversion. Furthermore, chain extensions were carried out that indicate a very high reinitiation efficiency (poly(DMAAm): from 10 500 to 97 500 g mol–1, PDI = 1.08; poly(DEAAm): from 8500 to 83 000 g mol–1, PDI = 1.13; poly(NIPAAm): from 9000 to 90 000 g mol–1, PDI = 1.11). The resulting polymers were thoroughly characterized via N,N-dimethylacetamide (DMAc) size exclusion chromatography, 1H NMR, and electrospray ionization-mass spectrometry (ESI-MS).
Co-reporter:Mathias Glassner, Kim K. Oehlenschlaeger, Till Gruendling, and Christopher Barner-Kowollik
Macromolecules 2011 Volume 44(Issue 12) pp:4681-4689
Publication Date(Web):May 31, 2011
DOI:10.1021/ma2010052
A strategy for the modular ambient temperature synthesis of ABA and ABC triblock copolymers via a combination of photoinduced Diels–Alder reactions with thermal Diels–Alder reactions and azide–alkyne click chemistry is reported. Polystyrene (PS) and PMMA (PMMA) with α-2,5-dimethylbenzophenone and ω-cyclopentadienyl or ω-azide end-functionality were prepared via atom transfer radical polymerization (ATRP) and subsequent transformation of the bromine end-group. The phototriggered conjugation reaction proceeds via an in situ formation of highly reactive o-quinodimethanes. Maleimide-capped poly(tert-butyl acrylate) obtained via ATRP was employed as dienophile. Alkyne and maleimide functionalized poly(ethylene glycol) (PEG) were synthesized by esterification of monomethoxy PEG. PtBA-b-PMMA-b-PtBA and PtBA-b-PS-b-PtBA were successfully prepared in a one-pot reaction at ambient temperature combining photoinduced and thermal Diels–Alder reactions. ABC triblock copolymers (PtBA-b-PS-b-PEG) with narrow polydispersities were obtained via the combination of photoinduced Diels–Alder reactions with thermal Diels–Alder reactions as well as CuAAc chemistry. The polymers were characterized by size exclusion chromatography and 1H NMR spectroscopy.
Co-reporter:Anja S. Goldmann, Thomas Tischer, Leonie Barner, Michael Bruns, and Christopher Barner-Kowollik
Biomacromolecules 2011 Volume 12(Issue 4) pp:
Publication Date(Web):March 2, 2011
DOI:10.1021/bm101461h
A combination of reversible addition−fragmentation chain transfer (RAFT) polymerization and hetero Diels−Alder (HDA) cycloaddition was used to effect, under mild (T ≈ 20 °C), fast, and modular conditions, the grafting of poly(isobornyl acrylate) (Mn = 9800 g mol−1, PDI = 1.19) onto a solid cellulose substrate. The active hydroxyl groups expressed on the cellulose fibers were converted to tosylate leaving groups, which were subsequently substituted by a highly reactive cyclopentadienyl functionality (Cp). By employing the reactive Cp-functionality as a diene, thiocarbonyl thio-capped poly(isobornyl acrylate) synthesized via RAFT polymerization (mediated by benzyl pyridine-2-yldithioformiate (BPDF)) was attached to the surface under ambient conditions by an HDA cycloaddition (reaction time: 15 h). The surface-modified cellulose samples were analyzed in-depth by X-ray photoelectron spectroscopy, scanning electron microscopy, elemental analysis, Fourier transform infrared (FT-IR) spectroscopy as well as Fourier transform infrared microscopy employing a focal plane array detector for imaging purposes. The analytical results provide strong evidence that the reaction of suitable dienophiles with Cp-functional cellulose proceeds under mild reaction conditions (T ≈ 20 °C) in an efficient fashion. In particular, the visualization of individual modified cellulose fibers via high-resolution FT-IR microscopy corroborates the homogeneous distribution of the polymer film on the cellulose fibers.
Co-reporter:Dominik Voll, Thomas Junkers, and Christopher Barner-Kowollik
Macromolecules 2011 Volume 44(Issue 8) pp:2542-2551
Publication Date(Web):March 30, 2011
DOI:10.1021/ma2001977
Photolytically generated radicals (at a wavelength of 351 nm) derived from the acetophenone-type photoinitiators benzoin (2-hydroxy-1,2-diphenylethanone) and 2,4,6-trimethylbenzoin (2-hydroxy-1-mesityl-2-phenylethanone, TMB) (specifically the benzoyl and mesitoyl radical) are quantified in their ability to serve as initiating species in methyl methacrylate (MMA), ethyl methacrylate (EMA), and butyl methacrylate (BMA) bulk free radical polymerizations under optimized conditions. Herein, 2,4,6-trimethylbenzoin is employed for the first time as photoinitiator in pulsed laser polymerizations (PLP) employing a high-frequency excimer laser, constituting a new source for mesitoyl radicals. The current work presents an improved method for quantifying radical efficiency of photoinitiation processes using coupled online size exclusion chromatography−electrospray ionization mass spectrometry (SEC/ESI-MS) to analyze the obtained polymers. Because of the occurrence of side reactions during the benzoin-initiated MMA polymerization, reduced laser energies (∼0.35 mJ/pulse) as well as low polymerization temperatures (∼−5 °C) were employed, which avoids side product formation. A plot of the ratio of benzoyl to mesitoyl (derived from 2,4,6-trimethylbenzoin) end groups vs the ratio of both initiators in the reaction mixture indicates that the benzoin-derived benzoyl radical is 3.0 (2.6, 2.4) times more likely to initiate the polymerization process of MMA (EMA, BMA) than the TMB-derived mesitoyl fragment. This observation is in sharp contrast to the case when mesitil is employed as a source of mesitoyl radicals (8.6 times higher likelihood of benzoyl incorporation). These results clearly support the notion that the origin of a radical species significantly determines its propensity to be incorporated at a polymer chain’s terminus. The cause of such an origin dependence is tentatively assigned—at least in part—to different triplet lifetimes or intersystem crossing efficiencies (ΦISC) or both of TMB and mesitil.
Co-reporter:Thomas Paulöhrl;Andrew J. Inglis
Advanced Materials 2010 Volume 22( Issue 25) pp:2788-2791
Publication Date(Web):
DOI:10.1002/adma.201000361
Co-reporter:Leena Nebhani;Detlef Schmiedl;Leonie Barner
Advanced Functional Materials 2010 Volume 20( Issue 12) pp:2010-2020
Publication Date(Web):
DOI:10.1002/adfm.200902330
Abstract
The surface modification of divinylbenzene (DVB)-based microspheres is performed via a combination of reversible addition fragmentation chain transfer (RAFT) polymerization and rapid hetero-Diels–Alder (HDA) chemistry with the aim of quantifying the grafting densities achieved using this “grafting-to” method. Two variants of the RAFT-HDA concept are employed to achieve the functionalization of the microspheres. In the first approach, the microspheres are functionalized with a highly reactive diene, i.e., cyclopentadiene, and are subsequently reacted with polystyrene chains (number-averaged molecular weight, Mn = 4200 g mol−1; polydispersity index, PDI = 1.12.) that carry a thiocarbonyl moiety functioning as a dienophile. The functionalization of the microspheres is achieved rapidly under ambient conditions, without the aid of an external catalyst. The surface grafting densities obtained are close to 1.2 × 1020 chains per gram of microspheres. In the second approach, the functionalization proceeds via the double bonds inherently available on the microspheres, which are reacted with poly(isobornyl acrylate) chains carrying a highly dienophilic thiocarbonyl functionality; two molecular weights (Mn = 6000 g mol−1, PDI = 1.25; Mn = 26 000 g mol−1, PDI = 1.26) are used. Due to the less reactive nature of the dienes in the second approach, functionalization is carried out at elevated temperatures (T = 60 °C) yet in the absence of a catalyst. In this case the surface grafting density is close to 7 chains nm−2 for Mn = 6000 g mol−1 and 4 chains nm−2 for Mn = 26 000 g mol−1, or 2.82 × 1019 and 1.38 × 1019 chains g−1, respectively. The characterization of the microspheres at various functionalization stages is performed via elemental analysis for the quantification of the grafting densities and attenuated total reflectance (ATR) IR spectroscopy as well as confocal microscopy for the analysis of the surface chemistry.
Co-reporter:Ozcan Altintas, Peter Gerstel, Nico Dingenouts and Christopher Barner-Kowollik
Chemical Communications 2010 vol. 46(Issue 34) pp:6291-6293
Publication Date(Web):29 Jul 2010
DOI:10.1039/C0CC00702A
α,ω-Hydrogen donor/acceptor functional polymer strands are prepared via a combination of living radical polymerization and orthogonal conjugation and subsequently self-assembled as single chains to emulate—on a simple level—the self-folding behaviour of natural biomacromolecules.
Co-reporter:Edgar H. H. Wong, Cyrille Boyer, Martina H. Stenzel, Christopher Barner-Kowollik and Thomas Junkers
Chemical Communications 2010 vol. 46(Issue 11) pp:1959-1961
Publication Date(Web):14 Jan 2010
DOI:10.1039/B925390D
Nitrones are demonstrated to efficiently mediate radical coupling reactions on the example of the conjugation of ATRP-made polymers, yielding macromolecules with distinct functional alkoxyamine centres in mid-chain locations of the chains.
Co-reporter:Mathias Dietrich, Mathias Glassner, Till Gruendling, Christina Schmid, Jana Falkenhagen and Christopher Barner-Kowollik
Polymer Chemistry 2010 vol. 1(Issue 5) pp:634-644
Publication Date(Web):01 Mar 2010
DOI:10.1039/B9PY00273A
We report the systematic investigation of a recently introduced one-pot radical transformation of methacrylate and acrylate-type polymers prepared via reversible addition fragmentation chain transfer (RAFT) polymerization into hydroxyl functional polymers. The simple reaction procedure involves stirring a solution of the RAFT functional polymer and an azo-initiator in tetrahydrofuran at elevated temperatures (T = 60 °C) in the presence of ambient air. Subsequent reduction of the formed hydroperoxide functional polymers to hydroxyl functional polymers is achieved in a one-pot procedure using triphenylphosphine. Polymers investigated in the current study are poly(methyl acrylate) (pMA), poly(butyl acrylate) (pBA), poly(isobornyl acrylate) (piBoA) and poly(tert-butyl acrylate) (ptBA) carrying a dithiobenzoate or phenyldithioacetate end terminius as well as a symmetrical trithiocarbonate mid chain function. Quantitative conversion into the hydroperoxyl and hydroxyl terminated product is observed when trithiocarbonate functional polymers are employed. In the case of dithiobenzoate and phenyldithioacetate functional acrylic polymers, some minor side products due to the oxidation of the RAFT end-group are generated. Size exclusion chromatography (SEC) and size exclusion chromatography–electrospray mass spectrometry (SEC-ESI-MS) were employed to monitor the progress of the reaction and to investigate the proposed reaction mechanism for the model polymers. When trithiocarbonate functional polymers are employed in the transformation reaction, the SEC analysis shows a bisection of the initial Mn. Collision induced dissociation (CID) MS experiments of the intermediate reaction products were conducted to gain in-depth information about the chemical structure. The new backbone linked hydroxyl group provides a versatile anchor for chemical end-group conversions and conjugation reactions with RAFT prepared polymers, alleviating problems with the rather limited ability of the dithioester end-group to undergo non-radical transformations.
Co-reporter:Till Gruendling, Steffen Weidner, Jana Falkenhagen and Christopher Barner-Kowollik
Polymer Chemistry 2010 vol. 1(Issue 5) pp:599-617
Publication Date(Web):26 Jan 2010
DOI:10.1039/B9PY00347A
Two decades after the introduction of matrix assisted laser desorption/ionization (MALDI) and electrospray ionization (ESI), soft ionization mass spectrometry represents a powerful toolset for the structural investigation of synthetic polymers. The present review highlights the current state-of-the-art, covering the latest developments of novel techniques, enabling instrumentation as well as the important applications of soft ionization MS from the beginning of 2008. Special attention is paid to the role that soft ionization MS has played in the mechanistic investigation of radical polymerization processes since 2005.
Co-reporter:Christopher Barner-Kowollik, Francesca Bennet, Maria Schneider-Baumann, Dominik Voll, Thomas Rölle, Thomas Fäcke, Marc-Stephan Weiser, Friedrich-Karl Bruder and Thomas Junkers
Polymer Chemistry 2010 vol. 1(Issue 4) pp:470-479
Publication Date(Web):19 Jan 2010
DOI:10.1039/B9PY00352E
Temperature dependent propagation rate coefficients, kp, are determined for four acrylate monomers containing a carbamate moiety via the pulsed laser polymerization-size exclusion chromatography (PLP-SEC) technique. Therefore, the Mark–Houwink–Kuhn–Sakurada coefficients K and a of the respective polymers were additionally determined via triple-detection SEC. The monomers under investigation were synthesized from hydroxyethyl acrylate, hydroxyl(iso)propyl acrylate as well as phenyl isocyanate and hexyl isocyanate, respectively, in all four possible combinations. For 2-(phenylcarbamoyloxy)ethyl acrylate (PhCEA) an activation energy of 14.3 kJ mol−1 and a frequency factor of A = 1.2 × 107 L·mol−1 s−1 are obtained for kp. The MHKS parameters for poly(PhCEA) are K = 8.3 × 10−5 dL g−1 and a = 0.677. For 2-(phenylcarbamoyloxy)isopropyl acrylate (PhCPA) an activation energy of 14.2 kJ mol−1 and a frequency factor of A = 4.9 × 106 L mol−1 s−1 are found for kp and the MHKS parameters for poly(PhCPA) read K = 10.3 × 10−5 dL g−1 and a = 0.657. The activation parameters of kp of 2-(hexylcarbamoyloxy)ethyl acrylate (HCEA) are EA = 13.3 kJ mol−1 and A = 6.6 × 106 L mol−1 s−1 with K = 36.0 × 10−5 dL g−1 and a = 0.552 for poly(HCEA). For 2-(hexylcarbamoyloxy)isopropyl acrylate (HCPA) EA is 14.1 kJ mol−1 and A = 6.6 × 106 L mol−1 s−1 with K = 26.0 × 10−5 dL g−1 and a = 0.587 for poly(HCPA). All rate measurements were performed in 1 M solutions in butyl acetate. The fast propagating nature and reduced activation energy of the monomers may be understood on the basis of the increased nucleophilicity that is induced by the carbamate functionality present in all monomers. Rate-increasing effects from solvent polarity and/or from H-bonding can, however, not be excluded and might also contribute to the observed high propagation rates.
Co-reporter:Thomas Junkers, Sandy P. S. Koo and Christopher Barner-Kowollik
Polymer Chemistry 2010 vol. 1(Issue 4) pp:438-441
Publication Date(Web):04 Mar 2010
DOI:10.1039/C0PY00019A
For the first time, the propagation rate coefficient kp of acrylonitrile has been determined in a broad temperature range in propylene carbonate solution via the pulsed laser polymerization–size exclusion chromatography technique employing a high frequency pulsed laser system. kp was determined to fit the Arrhenius relation ln(kp/L mol−1 s−1) = 14.42 − 1855K/T.
Co-reporter:Leena Nebhani
Macromolecular Rapid Communications 2010 Volume 31( Issue 14) pp:1298-1305
Publication Date(Web):
DOI:10.1002/marc.201000142
Co-reporter:Andrew J. Inglis
Macromolecular Rapid Communications 2010 Volume 31( Issue 14) pp:1247-1266
Publication Date(Web):
DOI:10.1002/marc.200900924
Co-reporter:Andreas Kaiser;Sven Brau;Michael Klimpel
Macromolecular Rapid Communications 2010 Volume 31( Issue 18) pp:1616-1621
Publication Date(Web):
DOI:10.1002/marc.201000162
Co-reporter:Andrew J. Inglis, Philippe Pierrat, Thierry Muller, Stefan Bräse and Christopher Barner-Kowollik
Soft Matter 2010 vol. 6(Issue 1) pp:82-84
Publication Date(Web):30 Oct 2009
DOI:10.1039/B920806M
The use of a hexakisazido macrocyclic methanofullerene proves to be highly efficient in the preparation of 6-arm star polymers via copper(I) catalyzed azide-alkyne cycloaddition.
Co-reporter:Francesca Bennet;Gene Hart-Smith;Till Gruendling;Thomas P. Davis;Philip J. Barker
Macromolecular Chemistry and Physics 2010 Volume 211( Issue 10) pp:1083-1097
Publication Date(Web):
DOI:10.1002/macp.200900625
Co-reporter:Till Gruendling;Dominik Voll;Michael Guilhaus
Macromolecular Chemistry and Physics 2010 Volume 211( Issue 1) pp:80-90
Publication Date(Web):
DOI:10.1002/macp.200900394
Co-reporter:Francesca Bennet;Philip J. Barker;Thomas P. Davis;Alexer H. Soeriyadi
Macromolecular Chemistry and Physics 2010 Volume 211( Issue 18) pp:2034-2052
Publication Date(Web):
DOI:10.1002/macp.201000133
Co-reporter:Gene Hart-Smith
Macromolecular Chemistry and Physics 2010 Volume 211( Issue 14) pp:1507-1529
Publication Date(Web):
DOI:10.1002/macp.201000107
Co-reporter:Till Gruendling;Thomas Junkers;Michael Guilhaus
Macromolecular Chemistry and Physics 2010 Volume 211( Issue 5) pp:520-528
Publication Date(Web):
DOI:10.1002/macp.200900323
Co-reporter:Antoine Bousquet;Martina H. Stenzel
Journal of Polymer Science Part A: Polymer Chemistry 2010 Volume 48( Issue 8) pp:1773-1781
Publication Date(Web):
DOI:10.1002/pola.23943
Abstract
Comb polymers were synthesized by the “grafting-onto” method via a combination of Reversible Addition-Fragmentation Chain Transfer (RAFT) polymerization and the hetero-Diels-Alder (HDA) cycloaddition. The HDA reactive monomer trans, trans-hexa-2,4-dienylacrylate (ttHA) was copolymerized with styrene via the RAFT process. Crosslinking was minimized by decreasing the monomer concentration—whilst keeping monomer to polymer conversions low—resulting in reactive backbones with on average one reactive pendant diene groups for 10 styrene units. The HDA cycloaddition was performed between the diene functions of the copolymer and a poly(n-butyl acrylate) (PnBA) prepared via RAFT polymerization with pyridin-2-yldithioformate, which can act as a dienophile. The coupling reactions were performed within 24 h at 50 °C and the grafting yield varies from 75% to 100%, depending on the number average molecular weight of the PnBA (3500 g mol−1 < Mn < 13,000 g mol−1) grafted chain and the reaction stoichiometry. The molecular weights of the grafted block copolymers range from 19,000 g mol−1 to 58,000 g mol−1 with polydispersities close to 1.25. © 2010 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem 48: 1773–1781, 2010
Co-reporter:Leena Nebhani
Journal of Polymer Science Part A: Polymer Chemistry 2010 Volume 48( Issue 2) pp:
Publication Date(Web):
DOI:10.1002/pola.23798
No abstract is available for this article.
Co-reporter:Lin Zang, Edgar H.H. Wong, Christopher Barner-Kowollik, Thomas Junkers
Polymer 2010 Volume 51(Issue 17) pp:3821-3825
Publication Date(Web):4 August 2010
DOI:10.1016/j.polymer.2010.06.040
The nitrone mediated polymerization of methyl methacrylate (MMA) via the enhanced (termination) spin capturing polymerization (ESCP) process is made possible via the addition of small amounts of styrene (between 5 and 10 vol.%) to the reaction mixture. Efficient control over the molecular weight between 7000 and 57,000 g mol−1 (at 60 °C) yields macromolecules that feature a mid-chain alkoxyamine functionality and are rich in methyl methacrylate. The collated kinetic and molecular weight data allow for a deduction of the spin capturing constant, CSC, in the range between 0.15 and 0.30. During the ESCP process, the number average molecular weight, Mn, of the formed mid-chain functional polymer is constant up to high monomer to polymer conversions (i.e. 80%). The high degree of alkoxyamine mid-chain functionality present in the generated polymeric material is evidenced via a subsequent nitroxide-mediated polymerization process employing the formed ESCP polymer, indicating a chain extension from 37,700 to 118,000 g mol−1 with a concomitant reduction in polydispersity (from 2.3 to 1.5).
Co-reporter:Pierre-Eric Millard, Leonie Barner, Jürgen Reinhardt, Michael R. Buchmeiser, Christopher Barner-Kowollik, Axel H.E. Müller
Polymer 2010 Volume 51(Issue 19) pp:4319-4328
Publication Date(Web):3 September 2010
DOI:10.1016/j.polymer.2010.07.017
The ambient temperature (20 °C) reversible addition fragmentation chain transfer (RAFT) polymerization of several water-soluble monomers conducted directly in aqueous media under γ-initiation (at dose rates of 30 Gy h−1) proceeds in a controlled fashion. Using functional trithiocarbonates, i.e., S,S-bis(α,α′-dimethyl-α″-acetic acid) trithiocarbonate (TRITT), 3-benzylsulfanyl thiocarbonylsulfanyl propionic acid (BPATT), and dithioester, i.e., 4-cyanopentanoic acid dithiobenzoate (CPADB), as chain transfer agents, fully water-soluble polymers of monomers such as N,N-dimethylacrylamide, 2-hydroxyethyl acrylate, acrylamide or oligo(ethylene glycol) methacrylate and stimuli-responsive polymers of monomers such as acrylic acid, N -isopropylacrylamide, 2-(dimethylamino)ethyl methacrylate or 2-acrylamido-2-methylpropane sulfonic acid can be obtained over a wide range of degrees of polymerization up to 10,000 with low polydispersity (typically M¯w/M¯n<1.2) to near quantitative conversions. Well-defined block copolymers between these monomers, based on several asymmetric macro-RAFT agents, can be obtained, suggesting that the RAFT agents are stable throughout the polymerization process so that complex and well-defined architectures can be obtained.
Co-reporter:Marianne Gaborieau, Leena Nebhani, Robert Graf, Leonie Barner and Christopher Barner-Kowollik
Macromolecules 2010 Volume 43(Issue 8) pp:3868-3875
Publication Date(Web):March 16, 2010
DOI:10.1021/ma100149p
The development of a solid-state nuclear magnetic resonance (NMR) method allowing the quantification of active sites (i.e., residual vinyl groups) accessible for chemical functionalization on the surface of poly(divinyl benzene) microspheres is presented. Residual vinyl groups of poly(divinyl benzene) microspheres (PDVB55 and PDVB80) were quantified via solid-state 13C cross-polarization magic-angle spinning (CP-MAS) NMR spectroscopy. In addition, 13C CP-MAS NMR spectroscopy allows the comparison of core and grafted microspheres functionalization on the same (arbitrary) scale in a short measuring time. This scale was calibrated by an extended absolute quantification of the vinyl groups using 13C single pulse excitation (SPE) MAS NMR spectroscopy. The degree of cross-linking of the microspheres was calculated to be 30 and 50% for PDVB55 and PDVB80 microspheres, respectively. The number of active groups per nominal surface area is 110 and 179 groups per nm2 for PDVB55 and PDVB80 microspheres, respectively. The loading capacities of the microspheres (e.g., 0.61 and 0.65 mmol·g−1) are not too far removed from those found in Merrifield resins of comparable sizes.
Co-reporter:Edgar H. H. Wong, Martina H. Stenzel, Thomas Junkers and Christopher Barner-Kowollik
Macromolecules 2010 Volume 43(Issue 8) pp:3785-3793
Publication Date(Web):March 16, 2010
DOI:10.1021/ma100263k
A novel nitrone (α-4-(3-(trimethylsilyl)prop-2-ynyloxy)-N-tert-butyl nitrone) with an alkyne “click” function is synthesized and employed in enhanced spin capturing polymerization (ESCP) as well as in radical coupling reactions between polymers preformed by atom transfer radical polymerization (ATRP) to generate midchain functionalized polymers. Both techniques allow for the facile introduction of chemical functionalities into a polymer midchain position and hence provide an attractive synthetic avenue for the construction of complex macromolecular architectures. Such a strategy is evidenced by the efficient use of polystyrene and poly(isobornyl acrylate) featuring an alkoxyamine midchain function in polymer−polymer conjugation reactions with azide-terminated polymers, yielding 3-arm star (co)polymers via the Cu-catalyzed alkyne/azide cycloaddition reactions. The successful formation of star-block co- and homopolymers is confirmed by conventional size exclusion chromatography (SEC) as well as via hyphenated liquid absorption chromatography under critical conditions (LACCC)-SEC techniques. The synthetic approach reported herein demonstrates that click functional nitrones can efficiently be employed as macromolecular construction agents in modular reactions.
Co-reporter:Andrew J. Inglis, Thomas Paulöhrl and Christopher Barner-Kowollik
Macromolecules 2010 Volume 43(Issue 1) pp:33-36
Publication Date(Web):December 8, 2009
DOI:10.1021/ma902464a
Co-reporter:Thomas Junkers, Maria Schneider-Baumann, Sandy S. P. Koo, Patrice Castignolles, and Christopher Barner-Kowollik
Macromolecules 2010 Volume 43(Issue 24) pp:10427-10434
Publication Date(Web):December 7, 2010
DOI:10.1021/ma102130h
The radical propagation rate coefficients, kp, of methyl acrylate (MA) and 2-ethylhexyl acrylate (EHA) have been determined in bulk via high frequency (500 Hz) pulsed laser polymerization coupled to size exclusion chromatography (PLP−SEC) up to elevated temperatures (20 ≤ T/°C ≤ 80). Prior to the analysis of the generated polymeric material, an investigation into the branching behavior of the generated polymers has been undertaken, employing the concept of local dispersity, D(Ve). In addition, the Mark-Houwkink-Kuhn-Sakurada parameters for both poly(MA) and polyEHA were determined at each studied reaction temperature. The temperature averaged values read (K = 10.2 × 10−5 dL g−1; α = 0.741) and (K = 9.85 × 10−5 dL g−1; α = 0.719) for poly(MA) and polyEHA, respectively. The local dispersity data indicate that branching in polyEHA may be considerably more prevalent than in poly(MA), as with increasing temperature polymer microinhomogeneities are observed. Consequently, the Arrhenius parameters for kp of EHA are beset with a larger error than those of MA. The activation parameters in the temperature range between 20 and 80 °C read: EAMA = 18.5 (+0.8 to −0.9) kJ·mol−1 and AMA = 2.5 (+1.2 to −0.6) × 107 L·mol−1·s−1; EAEHA = 15.8 (+1.6 to −1.4) kJ·mol−1 and AEHA = 9.1 (+10.1 to −2.9) × 106 L·mol−1·s−1.
Co-reporter:Marianne Gaborieau, Sandy P. S. Koo, Patrice Castignolles, Thomas Junkers and Christopher Barner-Kowollik
Macromolecules 2010 Volume 43(Issue 13) pp:5492-5495
Publication Date(Web):June 11, 2010
DOI:10.1021/ma100991c
Co-reporter:Andrew J. Inglis, Leena Nebhani, Ozcan Altintas, Friedrich Georg Schmidt and Christopher Barner-Kowollik
Macromolecules 2010 Volume 43(Issue 13) pp:5515-5520
Publication Date(Web):June 10, 2010
DOI:10.1021/ma100945b
Co-reporter:Anna-Marie Zorn;Thomas Junkers
Macromolecular Rapid Communications 2009 Volume 30( Issue 23) pp:2028-2035
Publication Date(Web):
DOI:10.1002/marc.200900536
Co-reporter:Christopher Barner-Kowollik Guest Editor
Macromolecular Rapid Communications 2009 Volume 30( Issue 23) pp:1961-1963
Publication Date(Web):
DOI:10.1002/marc.200900676
Co-reporter:Bart Dervaux;Thomas Junkers;Filip E. Du Prez
Macromolecular Reaction Engineering 2009 Volume 3( Issue 9) pp:529-538
Publication Date(Web):
DOI:10.1002/mren.200900046
Co-reporter:Leena Nebhani;Peter Gerstel;Petia Atanasova;Michael Bruns
Journal of Polymer Science Part A: Polymer Chemistry 2009 Volume 47( Issue 24) pp:7090-7095
Publication Date(Web):
DOI:10.1002/pola.23756
Co-reporter:Christopher Barner-Kowollik
Angewandte Chemie 2009 Volume 121( Issue 49) pp:9386-9388
Publication Date(Web):
DOI:10.1002/ange.200905145
Co-reporter:Christopher Barner-Kowollik
Angewandte Chemie International Edition 2009 Volume 48( Issue 49) pp:9222-9224
Publication Date(Web):
DOI:10.1002/anie.200905145
Co-reporter:Gene Hart-Smith, Christopher Barner-Kowollik
Polymer 2009 50(22) pp: 5175-5180
Publication Date(Web):
DOI:10.1016/j.polymer.2009.09.022
Co-reporter:Anna-Marie Zorn ; Thomas Junkers
Macromolecules () pp:
Publication Date(Web):August 4, 2011
DOI:10.1021/ma201345m
The free radical copolymerization of macromonomers with low molecular weight monomers represents a versatile tool for the formation of statistical copolymers featuring pendant side chains. In the current study n-butyl acrylate macromonomer (BAMM) has been synthesized via high temperature acrylate synthesis in a one-pot–one-step procedure and copolymerized with benzyl acrylate (BzA) as a comonomer up to 40% conversion in a free radical copolymerization with 1,1′-azobis(isobutyronitrile) (AIBN) as a source of radicals. The copolymers poly(BAMM)-co-poly(BzA) are fully characterized via size exclusion chromatography (SEC), nuclear magnetic resonance spectroscopy (NMR), and liquid adsorption chromatography at critical conditions (LACCC). The achievable molecular weight of the synthesized poly(BAMM)-co-poly(BzA) lies between 8000 and 77 000 g mol–1 with a polydispersity of 1.30–2.12. Calculated copolymer compositions from the integrals of the specific resonances Har and CH2 of the BzA compared to CH2 of the BAMM have been subjected to a (terminal model) Mayo–Lewis analysis, resulting in estimated reactivity ratios at ∼40% conversion of rBzA = 2.46 and rBAMM = 1.79, indicating a copolymer composition of FBA < 0.65 for the copolymers derived from fBzA > 0.9 up to a copolymer composition of FBA > 0.9 for copolymers with a comonomer feed of fBzA < 0.6. The poly(BAMM)-co-poly(BzA) have been analyzed under critical conditions of n-butyl acrylate (BA) on a normal phase column to obtain an image of the generated poly(BAMM)-co-poly(BzA).
Co-reporter:Dominik Voll ; Dmytro Neshchadin ; Kai Hiltebrandt ; Georg Gescheidt
Macromolecules () pp:
Publication Date(Web):July 25, 2012
DOI:10.1021/ma301275b
The analysis of photo-initiated poly(methyl methacrylate) via electrospray ionization-mass spectrometry (ESI–MS) (synthesized by pulsed laser polymerization (PLP, at λ = 351 nm) of methyl methacrylate (MMA) and benzoin as photoinitiator at 6 mJ/pulse laser energy) evidences the presence of unidentified species. The determination of the origin of these species requires a detailed investigation via size exclusion chromatography-electrospray ionization-mass spectrometry (SEC/ESI–MS) and chemically induced dynamic nuclear polarization-nuclear magnetic resonance spectroscopy (CIDNP–NMR). It was found that post-irradiation of benzoin-initiated poly(methyl methacrylate) leads to α-cleavage of the benzoyl fragment leading to a sequence of cascade reactions, including the formation of an additional double bond within the polymer chain as evidenced via ESI–MS. Furthermore, the reaction products of the benzoyl radical post α-cleavage (e.g., benzaldehyde, phenyl methyl ketone, methyl formate, or methane) as well as the formed macroradical can be followed by CIDNP–NMR, which allows establishing a reaction mechanism for the UV-induced cleavage process. The study thus evidence that—if the integrity of UV initiated polymers is to be kept intact during their synthesis—very low irradiation energies need to be employed.
Co-reporter:Mathias Glassner, Kim K. Oehlenschlaeger, Alexander Welle, Michael Bruns and Christopher Barner-Kowollik
Chemical Communications 2013 - vol. 49(Issue 6) pp:NaN635-635
Publication Date(Web):2012/11/23
DOI:10.1039/C2CC37651B
An efficient method for polymer surface patterning via Diels–Alder trapping of photo-generated thioaldehydes is presented. It is demonstrated that thioaldehyde end-groups generated by photolysis of phenacyl sulfides can be quantitatively trapped with various dienes. Poly(ethylene glycol) is immobilized on a surface in a spatially controlled fashion via irradiation through a shadow mask.
Co-reporter:Lukas Stolzer, Alexander S. Quick, Doris Abt, Alexander Welle, Denys Naumenko, Marco Lazzarino, Martin Wegener, Christopher Barner-Kowollik and Ljiljana Fruk
Chemical Communications 2015 - vol. 51(Issue 16) pp:NaN3366-3366
Publication Date(Web):2015/01/20
DOI:10.1039/C4CC08880H
Photoreactive gold nanoparticles (NP) can be encoded in a spatially resolved fashion using direct laser writing techniques into variable patterns. The surface of the gold nanoparticles is imparted with photoreactivity by tethering photo-caged dienes (‘photoenols’), which are able to undergo a rapid Diels–Alder cycloaddition with surface anchored enes. Subsequent to surface encoding, the particles feature residual caged dienes, which can be reactivated for secondary surface encoding.
Co-reporter:Yuuki Sugawara, Nils Jasinski, Michael Kaupp, Alexander Welle, Nicolas Zydziak, Eva Blasco and Christopher Barner-Kowollik
Chemical Communications 2015 - vol. 51(Issue 65) pp:NaN13003-13003
Publication Date(Web):2015/07/09
DOI:10.1039/C5CC05507E
An efficient methodology for modular fullerene functionalization via the photo-induced nitrile imine-mediated tetrazole–ene cycloaddition (NITEC) is introduced. The versatility and platform character of the method is illustrated by the light-driven reaction of fullerenes with small molecule, polymeric and surface-immobilized tetrazoles. The efficient fullerene conjugation is evidenced via mass spectrometric techniques.
Co-reporter:Thomas Josse, Ozcan Altintas, Kim K. Oehlenschlaeger, Philippe Dubois, Pascal Gerbaux, Olivier Coulembier and Christopher Barner-Kowollik
Chemical Communications 2014 - vol. 50(Issue 16) pp:NaN2026-2026
Publication Date(Web):2013/12/16
DOI:10.1039/C3CC49067J
The light induced, catalyst-free ambient temperature preparation of macrocyclic aliphatic polyesters is pioneered. Based on the photo-induced Diels–Alder reaction of orthoquinodimethane and acrylate moieties, cyclic polyesters of high purity are readily synthesized. Considering the high tolerance to functional groups and the orthogonality of the ligation, the reported protocol can be easily transferred to a large range of polymers, complex topologies (tadpole, sun-shaped, jellyfish, etc.) and applications.
Co-reporter:Johannes Willenbacher, Bernhard V. K. J. Schmidt, David Schulze-Suenninghausen, Ozcan Altintas, Burkhard Luy, Guillaume Delaittre and Christopher Barner-Kowollik
Chemical Communications 2014 - vol. 50(Issue 53) pp:NaN7059-7059
Publication Date(Web):2014/05/13
DOI:10.1039/C4CC03218G
In the present communication we introduce a new platform technology for the reversible folding of single polymer chains in aqueous environment on the basis of cyclodextrin (CD) host–guest chemistry and controlled radical polymerization protocols. The single-chain folding of adamantyl-β-CD α-ω-functionalized poly(N,N-dimethylacrylamide) and its reversion at elevated temperatures were monitored by DLS and nuclear Overhauser enhancement spectroscopy (NOESY).
Co-reporter:Michael Kaupp, Kai Hiltebrandt, Vanessa Trouillet, Patrick Mueller, Alexander S. Quick, Martin Wegener and Christopher Barner-Kowollik
Chemical Communications 2016 - vol. 52(Issue 9) pp:NaN1978-1978
Publication Date(Web):2015/12/10
DOI:10.1039/C5CC09444E
A wavelength selective technique for light-induced network formation based on two photo-active moieties, namely ortho-methylbenzaldehyde and tetrazole is introduced. The network forming species are photo-reactive star polymers generated via reversible activation fragmentation chain transfer (RAFT) polymerization, allowing the network to be based on almost any vinylic monomer. Direct laser writing (DLW) allows to form any complex three-dimensional structure based on the photo-reactive star polymers.
Co-reporter:Matthias Winkler, Lucas Montero de Espinosa, Christopher Barner-Kowollik and Michael A. R. Meier
Chemical Science (2010-Present) 2012 - vol. 3(Issue 8) pp:NaN2615-2615
Publication Date(Web):2012/05/16
DOI:10.1039/C2SC20402A
Heck coupling reactions are introduced as a modular, very efficient and highly orthogonal method for polymer–polymer conjugation. Several diblock and triblock copolymers (5200 Da ≤ Mn ≤ 17300 Da, 1.08 ≤ PDI ≤ 1.33) were prepared via Heck coupling reactions of acrylate-terminated and aryliodide-terminated polymers. The coupling reactions were performed using the so-called Jeffery's conditions, which allowed the use of equimolar amounts of reacting polymers and low reaction temperatures. Acrylated poly(ethylene glycol) monomethyl ether (PEG), poly(ε-caprolactone) (PCL) and polymers synthesized via head-to-tail selective acyclic diene metathesis (ADMET) polymerisation have been successfully conjugated with both a PEG and PCL containing an aryliodide moiety.
Co-reporter:Ozcan Altintas, Peter Gerstel, Nico Dingenouts and Christopher Barner-Kowollik
Chemical Communications 2010 - vol. 46(Issue 34) pp:NaN6293-6293
Publication Date(Web):2010/07/29
DOI:10.1039/C0CC00702A
α,ω-Hydrogen donor/acceptor functional polymer strands are prepared via a combination of living radical polymerization and orthogonal conjugation and subsequently self-assembled as single chains to emulate—on a simple level—the self-folding behaviour of natural biomacromolecules.
Co-reporter:Tanja K. Claus, Siham Telitel, Alexander Welle, Martin Bastmeyer, Andrew P. Vogt, Guillaume Delaittre and Christopher Barner-Kowollik
Chemical Communications 2017 - vol. 53(Issue 10) pp:NaN1602-1602
Publication Date(Web):2017/01/12
DOI:10.1039/C6CC09897E
We introduce a methodology to reversibly pattern planar surfaces via the light-induced dimerization of anthracenes, particularly involving a 9-triazolylanthracene motif. Specifically, we demonstrate that the visible light-induced forward reaction can be employed to pattern small molecule species as well as polymers in a spatially resolved fashion. Under UV irradiation, the generated patterns can be erased to regenerate reactive areas, which are then available for a new functionalization step. The dynamic change in surface chemistry is evidenced by ToF-SIMS.
Co-reporter:Mirela Zamfir, Cesar Rodriguez-Emmenegger, Stella Bauer, Leonie Barner, Axel Rosenhahn and Christopher Barner-Kowollik
Journal of Materials Chemistry A 2013 - vol. 1(Issue 44) pp:NaN6034-6034
Publication Date(Web):2013/08/19
DOI:10.1039/C3TB20880J
The reversible addition–fragmentation chain transfer polymerization of 2-hydroxyethyl methacrylate (HEMA) from surfaces (S-RAFT) using an R-group-attached chain transfer agent (CTA) is presented. The approach was exploited for the efficient preparation of well-defined PHEMA brushes of up to 50 nm thickness in a controlled fashion without using any cytotoxic catalyst. The chemical composition, morphology and wettability of the samples were assessed by X-ray photoelectron spectroscopy, atomic force microscopy and water contact angle measurements, while the growth kinetics were studied by monitoring the dry thickness via spectroscopic ellipsometry. The mechanism and kinetics of the RAFT polymerization on the surface – in the presence of a sacrificial CTA and of solvent mixtures with different polarities – were investigated. A marked effect of the concentration of the sacrificial CTA on the kinetics was observed. Importantly – and for the first time – the living PHEMA brushes were exploited as macroRAFT agents for chain extension, and thicknesses up to 70 nm were achieved. The prepared PHEMA brushes were challenged with protein solutions demonstrating their resistance to fouling.
Co-reporter:Daniel Volz, Astrid F. Hirschbiel, Daniel M. Zink, Jana Friedrichs, Martin Nieger, Thomas Baumann, Stefan Bräse and Christopher Barner-Kowollik
Journal of Materials Chemistry A 2014 - vol. 2(Issue 8) pp:NaN1462-1462
Publication Date(Web):2013/12/04
DOI:10.1039/C3TC32347A
The photoluminescence quantum efficiency as well as the processing properties of a series of brightly luminescent Cu(I)-metallopolymers strongly depended on the chosen synthetic approach. A monomeric, substituted styrenic complex features a photoluminescence quantum efficiency (PLQY) of only 4%, while its metallopolymeric thin film is over one order of magnitude more efficient.
Co-reporter:Daniel Volz, Thomas Baumann, Harald Flügge, Mathias Mydlak, Tobias Grab, Michael Bächle, Christopher Barner-Kowollik and Stefan Bräse
Journal of Materials Chemistry A 2012 - vol. 22(Issue 38) pp:NaN20790-20790
Publication Date(Web):2012/09/03
DOI:10.1039/C2JM33291D
The production of solution-processed OLEDs requires materials suitable for subsequent multilayer deposition. In the current study, we present an autocatalytic method to crosslink a luminescent copper(I)-complex with a polymeric backbone, in which the emitter itself acts as catalyst. In a showcase reaction demonstrating this concept for the first time, we combined a highly luminescent binuclear copper(I)-complex with a polystyrene derivative in order to prove the potential of the protocol. The luminescence properties were only slightly affected by the crosslinking, while the general stability increased drastically, as proven by thermogravimetric analysis (TGA). OLED tests confirmed the fundamental suitability of the concept for device applications as well as for subsequent solution-based multilayer deposition.
Co-reporter:Lebohang Hlalele ; Christoph J. Dürr ; Paul Lederhose ; Andreas Kaiser ; Stefan Hüsgen ; Sven Brandau
Macromolecules () pp:
Publication Date(Web):March 5, 2013
DOI:10.1021/ma4000896
An in-depth mechanistic study into the solution based initiator-free UV-induced radical copolymerization of 1,3-butadiene with acrylonitrile is reported. The light induced constant radical flux leads to moderate monomer conversions within 4 to 24 h. The number-average molecular weights of the prepared nitrile butadiene rubber (NBR) range from 2500 to 50 000 g mol–1 (1.7 ≤ PDI ≤ 2.4), while the achievable monomer conversion ranged from close to 7 up to 31% depending on the polymerization temperature, reaction time and UV light intensity. The rate coefficient for the generation of primary radicals, determined as the coupled parameter k1*k3, showed a dependence on the UV light intensity with values between 6.0 s–2 and 34.6 s–2 deduced for the UV light intensity range of 280 to 700 W. The estimated values of the lower limit average termination rate coefficient displayed no dependence on the UV light intensity, with lower limit values between 2.6 × 108 L mol–1 s–1 and 6.3 × 108 L mol–1 s–1 for the UV light intensity range of 280 to 700 W. The deduced values for the average termination rate coefficient were above the expected values for comparable average termination rate coefficients.
Co-reporter:Christoph J. Dürr ; Lebohang Hlalele ; Andreas Kaiser ; Sven Brandau
Macromolecules () pp:
Publication Date(Web):December 27, 2012
DOI:10.1021/ma302017c
An efficient approach for the synthesis of block copolymers of poly(acrylonitrile-co-butadiene) (NBR) and poly(styrene-co-acrylonitrile) (SAN) is described. Conjugation of preformed polymer building blocks is achieved via a hetero-Diels-Alder (HDA) mechanism employing cyclopentadiene-capped NBRs with dienophile SAN copolymers, both synthesized via reversible addition–fragmentation chain transfer (RAFT) polymerization. The protocol is further extended toward the synthesis of 4-miktoarm star polymers, consisting of two NBR and two SAN arms. Molar masses of the obtained complex macromolecular architectures range from below 10 000 g·mol–1 up to 110 000 g·mol–1 with dispersities below 1.5. Molecular verification of the coupling moieties is provided via NMR spectroscopy as well as ESI mass spectrometry. Size exclusion chromatography (SEC) traces of the obtained block copolymers and miktoarm star polymers were analyzed via deconvolution techniques, revealing the presence of 9.9–12.6 wt % (block copolymers) and 20 wt % (stars) of polymer chains not participating in the HDA conjugation, respectively. The residual polymers were analyzed toward their origin from either the loss of functionality during RAFT polymerization or incomplete conversion during the conjugation process. The comprehensive analysis of the macromolecular material was underpinned by kinetic simulations to estimate the fractions of nonfunctional polymer chains generated during the NBR and SAN polymerizations. The simulations evidenced that NBR-b-SAN samples cannot contain more than 94.4 wt % (Mn 13 000 g·mol–1), 93.6 wt % (Mn 57 000 g·mol–1), or 93.9 wt % (Mn 110 000 g·mol–1) of polymer chains actually possessing the targeted block copolymer structures when assuming an ideal RAFT process. These results unambiguously reveal that nonfunctionalized polymer chains formed during RAFT polymerization cause the incomplete conjugation of polymer building blocks, evidencing the limitations of end-group control in controlled/living radical polymerizations.
Co-reporter:Nils Wedler-Jasinski, Nicolas Delbosc, Marie-Alice Virolleaud, Damien Montarnal, Alexander Welle, Leonie Barner, Andreas Walther, Julien Bernard and Christopher Barner-Kowollik
Chemical Communications 2016 - vol. 52(Issue 56) pp:NaN8756-8756
Publication Date(Web):2016/06/20
DOI:10.1039/C6CC03612K
We introduce recodable surfaces solely based on reversible artificial hydrogen bonding interactions. We show that a symmetrical oligoamide (SOA) attached to poly(methyl methacrylate) (PMMA) can be repeatedly immobilized and cleaved off spatially defined surface domains photochemically functionalized with asymmetric oligoamides (AOAs). The spatially resolved recodability is imaged and quantified via ToF-SIMS.
Co-reporter:Kai Hiltebrandt, Michael Kaupp, Edgar Molle, Jan P. Menzel, James P. Blinco and Christopher Barner-Kowollik
Chemical Communications 2016 - vol. 52(Issue 60) pp:NaN9429-9429
Publication Date(Web):2016/06/28
DOI:10.1039/C6CC03848D
We introduce a light induced sequence enabling λ-orthogonal star polymer formation via an arms-first approach, based on an α,ω-functional polymer carrying tetrazole and o-methyl benzaldehyde moieties, which upon irradiation can readily undergo cycloaddition with a trifunctional maleimide core. Depending on the wavelength, the telechelic strand can be attached to the core at either photo-reactive end.
Co-reporter:Nicolas Zydziak, Florian Feist, Birgit Huber, Jan O. Mueller and Christopher Barner-Kowollik
Chemical Communications 2015 - vol. 51(Issue 10) pp:NaN1802-1802
Publication Date(Web):2014/12/03
DOI:10.1039/C4CC08756A
We report the first photochemical protocol for the generation of sequence defined macromolecules employing two hetero bifunctional photoreactive synthons, exploiting the orthogonal nature of photochemical – via the use of caged dienes – and thermally driven ligation protocols. We demonstrate that the iterative alternating synthon addition to an initial bifunctional core under irradiation at ambient temperature enables the generation of a macromolecule with up to 10 units (M = 3231.58 g mol−1, Đ = 1.00). The resulting macromolecules are monodisperse and feature absolute chain end fidelity. The unit-by-unit construction of the macromolecule is evidenced by Nuclear Magnetic Resonance Spectroscopy, Electrospray Ionization Mass Spectrometry and Size Exclusion Chromatography. The fundamental principle demonstrated herein paves the way for employing photochemical strategies for the design of sequence defined polymers.
Co-reporter:Jan O. Mueller, Dominik Voll, Friedrich G. Schmidt, Guillaume Delaittre and Christopher Barner-Kowollik
Chemical Communications 2014 - vol. 50(Issue 99) pp:NaN15684-15684
Publication Date(Web):2014/10/22
DOI:10.1039/C4CC07792J
A facile, fast and ambient-temperature avenue towards highly fluorescent polymers is introduced via polymerizing non-fluorescent photoreactive monomers based on light-induced NITEC chemistry, providing a platform technology for fluorescent polymers. The resulting polypyrazolines were analyzed in depth and the photo-triggered step-growth process was monitored in a detailed kinetic study.
Co-reporter:Basit Yameen, Cesar Rodriguez-Emmenegger, Ishtiaq Ahmed, Corinna M. Preuss, Christoph J. Dürr, Nicolas Zydziak, Vanessa Trouillet, Ljiljana Fruk and Christopher Barner-Kowollik
Chemical Communications 2013 - vol. 49(Issue 60) pp:NaN6736-6736
Publication Date(Web):2013/06/04
DOI:10.1039/C3CC43361G
An unprecedented one-pot procedure employing a cyclopentadienyl functionalized RAFT agent allowed the grafting of poly(carboxybetaine acrylamide) – a highly functional and biocompatible polymer – from the surface of pristine SWCNTs. The pendant carboxylic acid groups of the surface grafted polymer were further conjugated with single-stranded (ss)-DNA, which was successfully hybridized with a Cy5 labelled complementary DNA strand.
Co-reporter:Basit Yameen, Cesar Rodriguez-Emmenegger, Corinna M. Preuss, Ognen Pop-Georgievski, Elisseos Verveniotis, Vanessa Trouillet, Bohuslav Rezek and Christopher Barner-Kowollik
Chemical Communications 2013 - vol. 49(Issue 77) pp:NaN8625-8625
Publication Date(Web):2013/07/22
DOI:10.1039/C3CC44683B
Cyclopentadienyl end-capped poly(3-hexylthiophene) was employed to fabricate conductive surface tethered polymer brushes via a facile route based on cyclopentadiene–maleimide Diels–Alder ligation. The efficient nature of the Diels–Alder ligation was further combined with a biomimetic polydopamine-assisted functionalization of surfaces, making it an access route of choice for P3HT surface immobilization.
Co-reporter:Dennis M. Bauer, Anita Rogge, Lukas Stolzer, Christopher Barner-Kowollik and Ljiljana Fruk
Chemical Communications 2013 - vol. 49(Issue 77) pp:NaN8628-8628
Publication Date(Web):2013/07/31
DOI:10.1039/C3CC43291B
DNA was modified with a photo-reactive caged diene allowing for the modification of dienophile containing proteins under mild irradiation conditions to afford fully functional DNA–protein conjugates.
Co-reporter:Edgar H. H. Wong, Cyrille Boyer, Martina H. Stenzel, Christopher Barner-Kowollik and Thomas Junkers
Chemical Communications 2010 - vol. 46(Issue 11) pp:NaN1961-1961
Publication Date(Web):2010/01/14
DOI:10.1039/B925390D
Nitrones are demonstrated to efficiently mediate radical coupling reactions on the example of the conjugation of ATRP-made polymers, yielding macromolecules with distinct functional alkoxyamine centres in mid-chain locations of the chains.
Co-reporter:Edgar H. H. Wong, Ozcan Altintas, Martina H. Stenzel, Christopher Barner-Kowollik and Thomas Junkers
Chemical Communications 2011 - vol. 47(Issue 19) pp:NaN5493-5493
Publication Date(Web):2011/04/11
DOI:10.1039/C1CC10322A
A synthetic strategy employing nitrones as radical spin traps is presented on the example of the efficient generation of novel dendrimersvia a combination of radical and classical ‘click’ chemistry.
Co-reporter:Mathias Glassner, Kristian Kempe, Ulrich S. Schubert, Richard Hoogenboom and Christopher Barner-Kowollik
Chemical Communications 2011 - vol. 47(Issue 38) pp:NaN10622-10622
Publication Date(Web):2011/09/01
DOI:10.1039/C1CC14075B
An efficient method for the preparation of cyclopentadienyl endcapped poly(2-ethyl-2-oxazoline) (PEtOx–Cp) via cationic ring-opening polymerization utilizing sodium cyclopentadienide as a termination agent is presented. Subsequent Diels–Alder reactions with N-substituted maleimides proceed quantitatively at ambient temperature. A block copolymer (PEtOx-b-PEG) is prepared employing maleimide terminated poly(ethylene glycol).
Co-reporter:Martina M. Cecchini, Jan Steinkoenig, Samantha Reale, Leonie Barner, Jiayin Yuan, Anja S. Goldmann, Francesco De Angelis and Christopher Barner-Kowollik
Chemical Science (2010-Present) 2016 - vol. 7(Issue 8) pp:NaN4921-4921
Publication Date(Web):2016/04/21
DOI:10.1039/C6SC01347C
We introduce a universal high resolution mass spectrometric method for the analysis of poly(ionic liquid)s (PILs), which belong to the most challenging polyelectrolytes from an analytical perspective, by fusing high resolution collision-induced dissociation (CID)-Orbitrap mass spectrometry (MS) with supercharging agents as well as quadrupole time-of-flight (QToF) MS. The study includes a wide array of hydrophilic halide-containing PILs, which were analyzed in negative mode. The influence of the core structures (based on imidazolium, triazolium, ammonium, phosphonium and pyridinium moieties), and variable styrene-, acrylate- and vinyl-type IL polymers on the ionization behavior is mapped in detail. Variable end group functionalities were introduced via functional chain transfer agents (CTA) in reversible addition-fragmentation chain transfer (RAFT) polymerization to study their behavior during the MS analysis. Furthermore, the demanding class of vinylimidazolium halide IL polymers was investigated. The current contribution thus introduces a new analytical technology platform for an entire polymer class.
Co-reporter:Kai Pahnke, Josef Brandt, Ganna Gryn'ova, Peter Lindner, Ralf Schweins, Friedrich Georg Schmidt, Albena Lederer, Michelle L. Coote and Christopher Barner-Kowollik
Chemical Science (2010-Present) 2015 - vol. 6(Issue 2) pp:NaN1074-1074
Publication Date(Web):2014/11/03
DOI:10.1039/C4SC02908A
We report the investigation of fundamental entropic chain effects that enable the tuning of modular ligation chemistry – for example dynamic Diels–Alder (DA) reactions in materials applications – not only classically via the chemistry of the applied reaction sites, but also via the physical and steric properties of the molecules that are being joined. Having a substantial impact on the reaction equilibrium of the reversible ligation chemistry, these effects are important when transferring reactions from small molecule studies to larger or other entropically very dissimilar systems. The effects on the DA equilibrium and thus the temperature dependent degree of debonding (%debond) of different cyclopentadienyl (di-)functional poly(meth-)acrylate backbones (poly(methyl methacrylate), poly(iso-butyl methacrylate), poly(tert-butyl methacrylate), poly(iso-butyl acrylate), poly(n-butyl acrylate), poly(tert-butyl acrylate), poly(methyl acrylate) and poly(isobornyl acrylate)), linked via a difunctional cyanodithioester (CDTE) were examined via high temperature (HT) NMR spectroscopy as well as temperature dependent (TD) SEC measurements. A significant impact of not only chain mass and length with a difference in the degree of debonding of up to 30% for different lengths of macromonomers of the same polymer type but – remarkably – as well the chain stiffness with a difference in bonding degrees of nearly 20% for isomeric poly(butyl acrylates) is found. The results were predicted, reproduced and interpreted via quantum chemical calculations, leading to a better understanding of the underlying entropic principles.
Co-reporter:Thomas Pauloehrl, Alexander Welle, Kim K. Oehlenschlaeger and Christopher Barner-Kowollik
Chemical Science (2010-Present) 2013 - vol. 4(Issue 9) pp:NaN3507-3507
Publication Date(Web):2013/06/17
DOI:10.1039/C3SC50815C
A novel and very simple photochemical strategy that utilizes light as a facile means to provide spatio-temporal control for the direct covalent immobilization of nucleophiles is presented. The concept is based upon the efficient trapping of photogenerated thioaldehydes by amines, hydroxylamines, and thiols. Surface patterns of polymers and small molecules bearing pendant amine-, hydroxylamine- or thiol moieties were successfully generated and imaged in a time-of-flight secondary-ion mass spectrometry (ToF-SIMS) investigation.
Co-reporter:Antonina Vigovskaya, Doris Abt, Ishtiaq Ahmed, Christof M. Niemeyer, Christopher Barner-Kowollik and Ljiljana Fruk
Journal of Materials Chemistry A 2016 - vol. 4(Issue 3) pp:NaN449-449
Publication Date(Web):2015/12/01
DOI:10.1039/C5TB02207J
A photocaged diene is introduced at the 5′-end of oligonucleotides using the H-phosphonate approach. The photoenol-functionalized DNA is subsequently employed for the conjugation to a protein and the spatially controlled immobilization onto surfaces using a light-induced Diels–Alder cycloaddition. Fully functional protein–DNA conjugates and patterned DNA surfaces are obtained under mild irradiation conditions.
Co-reporter:Corinna M. Preuss, Thomas Tischer, Cesar Rodriguez-Emmenegger, Markus M. Zieger, Michael Bruns, Anja S. Goldmann and Christopher Barner-Kowollik
Journal of Materials Chemistry A 2014 - vol. 2(Issue 1) pp:NaN40-40
Publication Date(Web):2013/10/22
DOI:10.1039/C3TB21317J
An avenue for the development of spatially resolved functional interfaces is presented. By introducing a novel, photo-reactive molecule – carrying a DOPA functionality and a photo-reactive group – we merge the ability of mussels to adhere to any surface with the spatial and temporal control of photo-click reactions, opening a plethora of applications in the biomedical and materials fields.
Co-reporter:Andrew P. Vogt, Julien De Winter, Peter Krolla-Sidenstein, Udo Geckle, Olivier Coulembier and Christopher Barner-Kowollik
Journal of Materials Chemistry A 2014 - vol. 2(Issue 23) pp:NaN3581-3581
Publication Date(Web):2014/04/04
DOI:10.1039/C4TB00491D
A degradable polyphthalaldehyde-polystyrene block copolymer generated by modular ligation is reported for the first time serving as a nanochannel template for the formation of nanostructured materials. The polyphthalaldehyde-b-polystyrene copolymer was spin-coated onto a surface with subsequent polyphthalaldehyde block removal. Block conjugation and block removal were confirmed by H-NMR, SEC, AFM, and SEM.
Co-reporter:Nathalie K. Guimard, Junming Ho, Josef Brandt, Ching Yeh Lin, Mansoor Namazian, Jan O. Mueller, Kim K. Oehlenschlaeger, Stefan Hilf, Albena Lederer, Friedrich G. Schmidt, Michelle L. Coote and Christopher Barner-Kowollik
Chemical Science (2010-Present) 2013 - vol. 4(Issue 7) pp:NaN2759-2759
Publication Date(Web):2013/05/02
DOI:10.1039/C3SC50642H
The widely accepted approach for controlling polymer debonding/rebonding properties in responsive materials has been to purposefully engineer the functional end-groups responsible for monomer dynamic bonding. Here, however, we evidence that the debonding temperature of a polymer can also be tuned by changing the chain length of the polymer building blocks, thus altering the entropy released on debonding. Entropy driven debonding, as governed by building block chain length, is suggested theoretically and realized experimentally for two Diels–Alder polymer systems, each based on a different difunctional diene and a common difunctional dienophile. In each case a significant decrease (as much as 60 °C) in the retro Diels–Alder temperature was observed when the chain length of the difunctional dienophile building block was increased. These results have the potential to fundamentally change the approach utilized to design materials capable of bonding reversibly on demand.
Co-reporter:Alexander P. Haehnel ; Maria Schneider-Baumann ; Kai U. Hiltebrandt ; Andrea M. Misske
Macromolecules () pp:
Publication Date(Web):December 18, 2012
DOI:10.1021/ma302319z
The Arrhenius parameters of the propagation rate coefficient for two linear methacrylates, two branched methacrylates, and two branched acrylates are determined via the pulsed laser polymerization–size exclusion chromatography (PLP-SEC) method. The Mark–Houwink–Kuhn–Sakurada parameters of these polymers are additionally determined via multidetector SEC of narrowly distributed polymer samples obtained through fractionation, allowing for a correct SEC calibration in the PLP-SEC experiment. The data obtained for stearyl methacrylate (SMA, A = 3.45 (−1.17 to +4.46) × 106 L·mol–1·s–1; Ea = 21.49 (−1.59 to +1.90) kJ·mol–1) and behenyl methacrylate (BeMA, A = 2.51 (−0.80 to +3.06) × 106 L·mol–1·s–1; Ea = 20.52 (−1.43 to +1.85) kJ·mol–1) underpin the trend of increasing kp with increasing ester side chain length. Propylheptyl methacrylate (PHMA, A = 2.83 (−0.82 to 3.15) × 106 L·mol–1·s–1; Ea = 21.72 (−1.20 to +1.64) kJ·mol–1) and heptadecanyl methacrylate (C17MA, A = 2.04 (−0.66 to +1.71) × 106 L·mol–1·s–1; Ea = 20.72 (−1.42 to +1.38) kJ·mol–1) can be described as a family of branched methacrylates jointly with isodecyl methacrylate and ethylhexyl methacrylate (both published previously), resulting in joint Arrhenius parameters of A = 2.39 (−0.51 to +0.84) × 106 L·mol–1·s–1 and Ea = 21.16 (−0.78 to +0.76) kJ·mol–1. In addition, the corresponding branched acrylates are studied applying high-frequency PLP at a 500 Hz laser repetition rate, resulting in Arrhenius parameters of A = 1.05 (−0.42 to +2.81) × 107 L·mol–1·s–1 and Ea = 16.41 (−1.99 to +2.42) kJ·mol–1 for propylheptyl acrylate (PHA) and A = 8.15 (−2.83 to +10.3) × 106 L·mol–1·s–1 and Ea = 14.66 (−1.49 to +1.66) kJ·mol–1 for heptadecanyl acrylate (C17A).

![Propanoic acid, 3-[[[(phenylmethyl)thio]thioxomethyl]thio]-, 2,2-bis[[1-oxo-3-[[[(phenylmethyl)thio]thioxomethyl]thio]propoxy]methyl]butyl ester](/data/chemimg/1361400/874747-27-0.png)
![Propanoic acid, 3-[[[(phenylmethyl)thio]thioxomethyl]thio]-, 2,2-bis[[1-oxo-3-[[[(phenylmethyl)thio]thioxomethyl]thio]propoxy]methyl]butyl ester](/data/chemimg/1361400/874747-27-0_b.png)
![Ferrocene, 1,1'-bis[(2-propyn-1-yloxy)methyl]-](/data/chemimg/3723400/1226860-94-1.png)
![Ferrocene, 1,1'-bis[(2-propyn-1-yloxy)methyl]-](/data/chemimg/3723400/1226860-94-1_b.png)
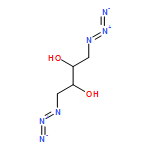
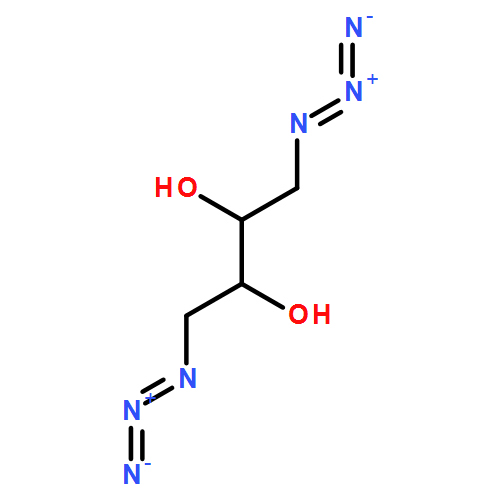



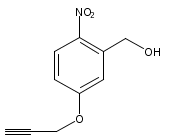




![Propanamide, 2-bromo-2-methyl-N-[3-(triethoxysilyl)propyl]-](/data/chemimg/107500/908595-77-7.png)
![Propanamide, 2-bromo-2-methyl-N-[3-(triethoxysilyl)propyl]-](/data/chemimg/107500/908595-77-7_b.png)


![Propanoic acid, 2-[[[(2-carboxyethyl)thio]thioxomethyl]thio]-2-methyl-](/data/chemimg/3502700/870451-10-8.png)
![Propanoic acid, 2-[[[(2-carboxyethyl)thio]thioxomethyl]thio]-2-methyl-](/data/chemimg/3502700/870451-10-8_b.png)
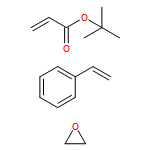
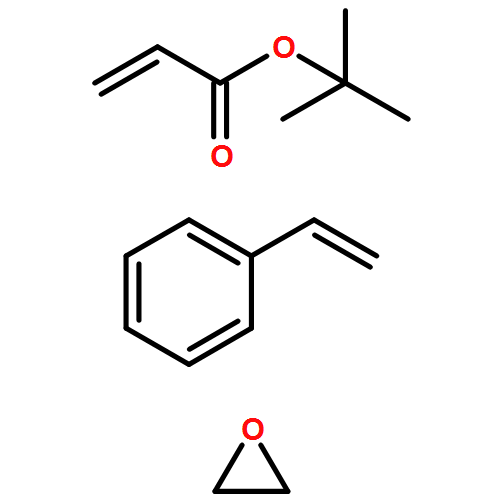
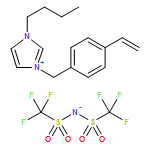
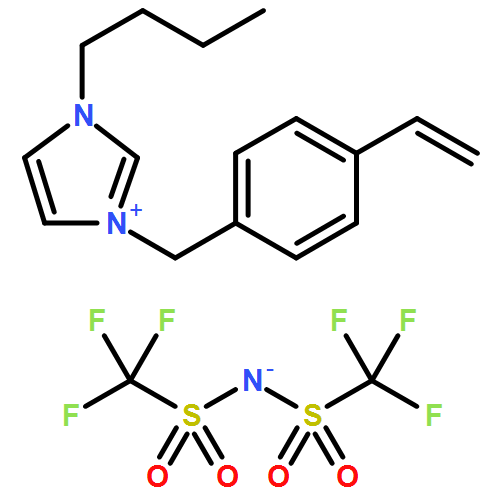
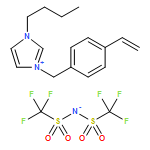
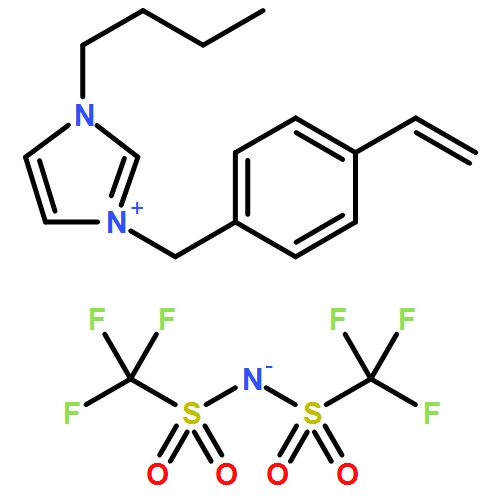
![Propanamide, 2-bromo-2-methyl-N-[3-(trihydroxysilyl)propyl]-](/data/chemimg/132400/855250-76-9.png)
![Propanamide, 2-bromo-2-methyl-N-[3-(trihydroxysilyl)propyl]-](/data/chemimg/132400/855250-76-9_b.png)
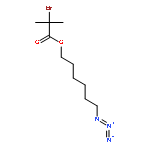





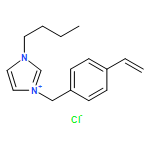
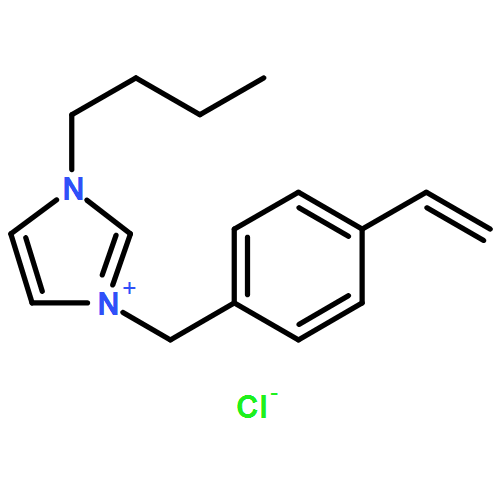







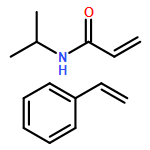
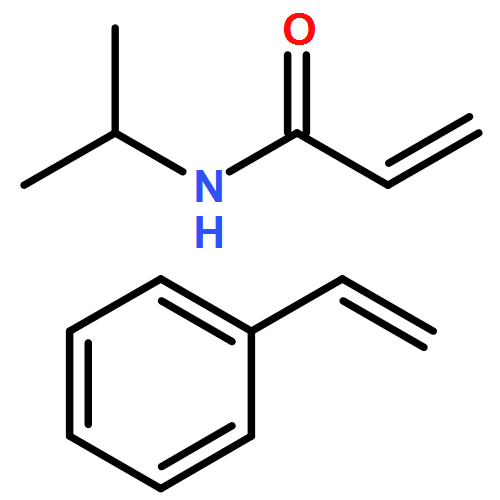










![Diazene, bis[4-[(trimethylsilyl)ethynyl]phenyl]-, (1E)-](http://img.cochemist.com/ccimg/269800/269747-83-3.png)
![Diazene, bis[4-[(trimethylsilyl)ethynyl]phenyl]-, (1E)-](http://img.cochemist.com/ccimg/269800/269747-83-3_b.png)

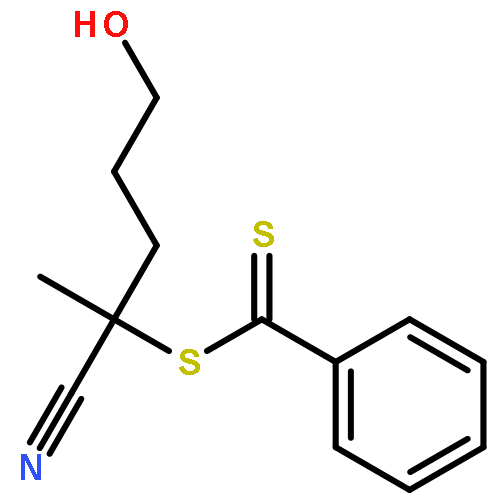
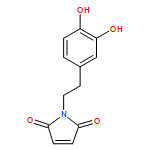
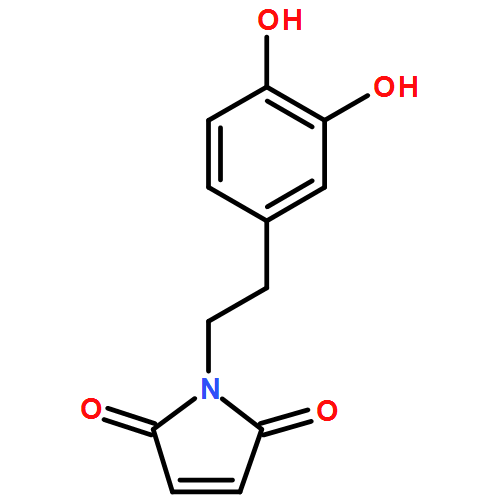


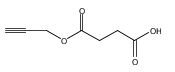
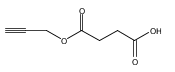
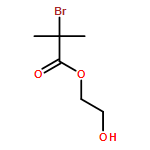
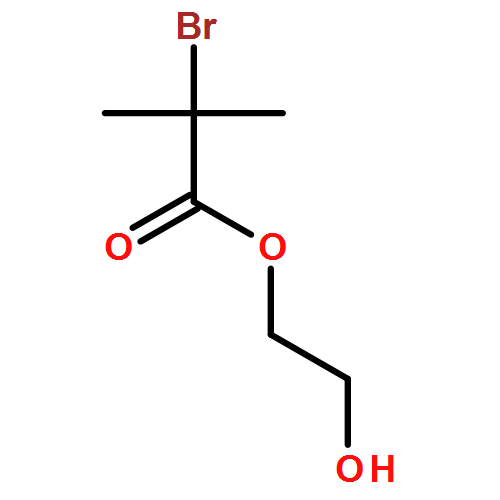
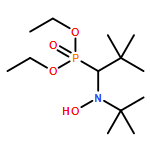
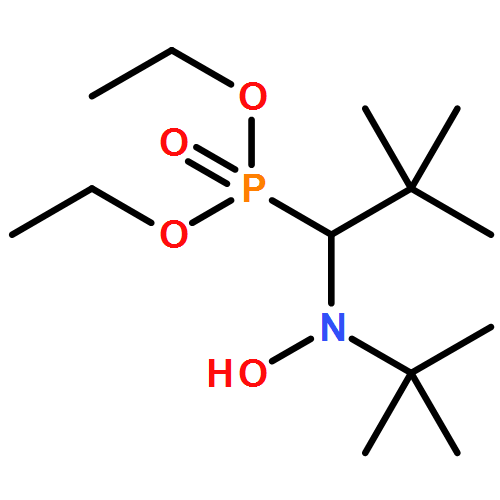
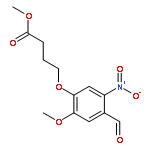
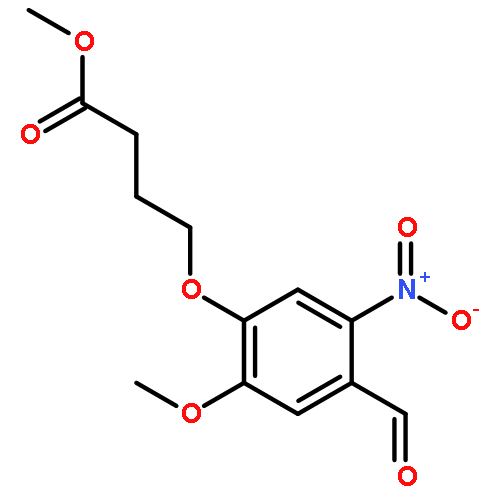
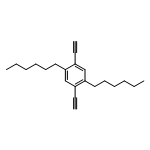

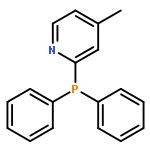
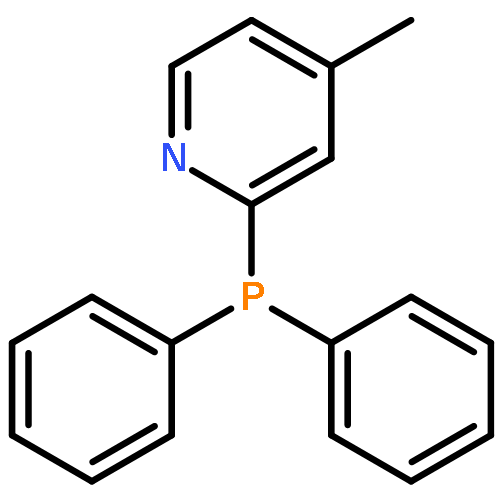
![[6'-acetyloxy-5-(2,5-dioxopyrrol-1-yl)-3-oxospiro[2-benzofuran-1,9'-xanthene]-3'-yl] Acetate](/data/chemimg/519900/150322-01-3.png)
![[6'-acetyloxy-5-(2,5-dioxopyrrol-1-yl)-3-oxospiro[2-benzofuran-1,9'-xanthene]-3'-yl] Acetate](/data/chemimg/519900/150322-01-3_b.png)








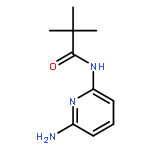
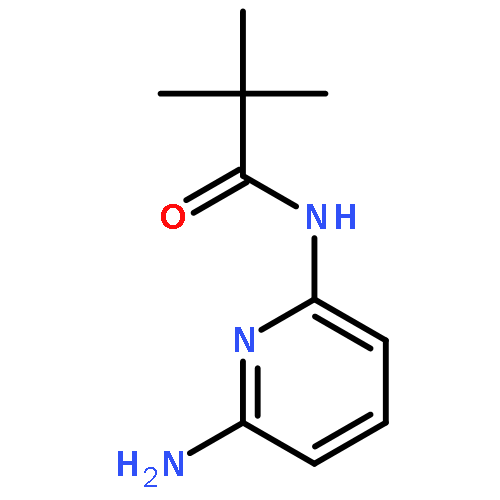
![Benzoic acid, 4-[[[(4-methylphenyl)sulfonyl]hydrazono]methyl]-, methylester](http://img.cochemist.com/ccimg/125600/125587-45-3.png)
![Benzoic acid, 4-[[[(4-methylphenyl)sulfonyl]hydrazono]methyl]-, methylester](http://img.cochemist.com/ccimg/125600/125587-45-3_b.png)








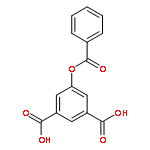
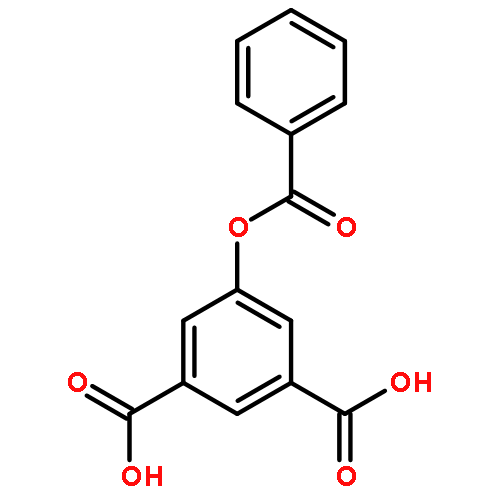
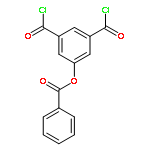
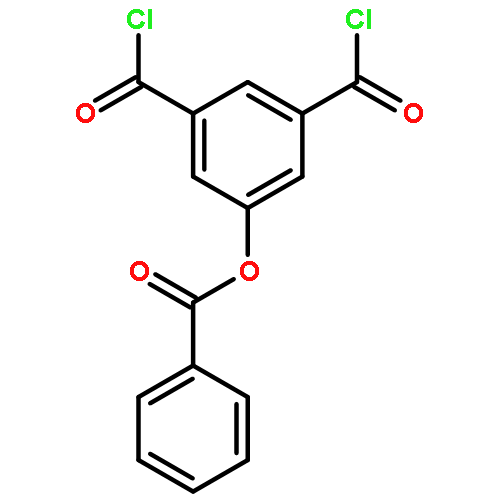


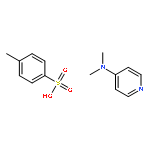

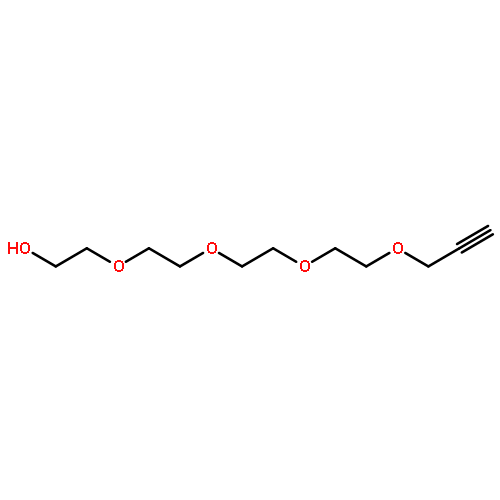
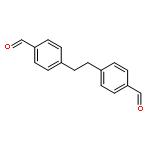
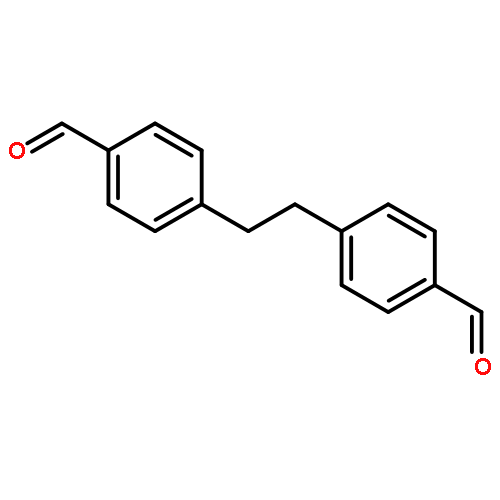

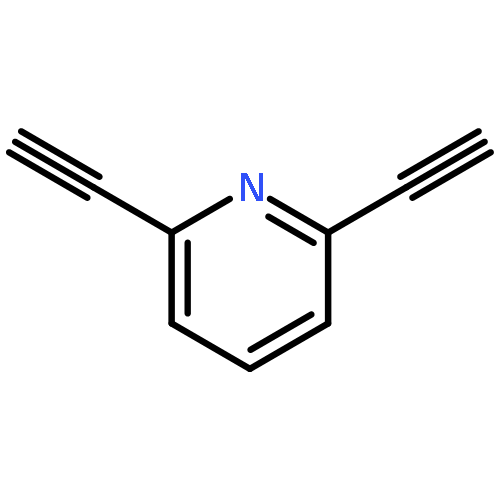
![N-BENZYL-N-[(2-CHLORO-8-METHYL-3-QUINOLINYL)METHYL]-2-FURAMIDE](http://img.cochemist.com/ccimg/75900/75867-44-6.png)
![N-BENZYL-N-[(2-CHLORO-8-METHYL-3-QUINOLINYL)METHYL]-2-FURAMIDE](http://img.cochemist.com/ccimg/75900/75867-44-6_b.png)
![POLY[[(1-OXOPROPYL)IMINO]-1,2-ETHANEDIYL]](http://img.cochemist.com/ccimg/69500/69488-61-5.png)
![POLY[[(1-OXOPROPYL)IMINO]-1,2-ETHANEDIYL]](http://img.cochemist.com/ccimg/69500/69488-61-5_b.png)
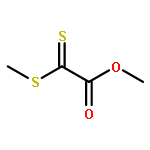
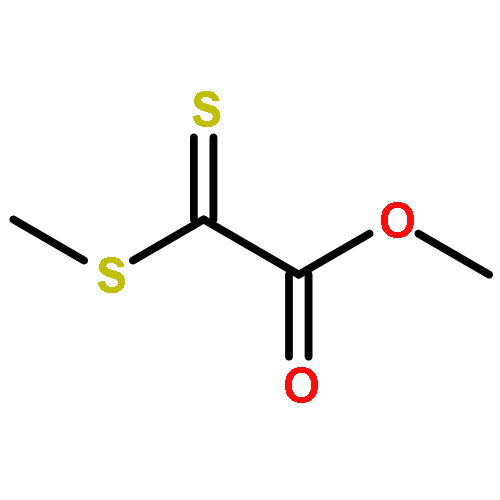
![Acetic acid, 2-[(2-oxo-2-phenylethyl)thio]-, ethyl ester](/data/chemimg/1141800/66560-19-8.png)
![Acetic acid, 2-[(2-oxo-2-phenylethyl)thio]-, ethyl ester](/data/chemimg/1141800/66560-19-8_b.png)
![2-PROPENOIC ACID, 2-METHYL-, 2-[(BROMOACETYL)OXY]ETHYL ESTER](http://img.cochemist.com/ccimg/65500/65462-76-2.png)
![2-PROPENOIC ACID, 2-METHYL-, 2-[(BROMOACETYL)OXY]ETHYL ESTER](http://img.cochemist.com/ccimg/65500/65462-76-2_b.png)
![2H-Cyclopenta[l]phenanthren-2-one, 1,3-bis(4-methoxyphenyl)-](http://img.cochemist.com/ccimg/62400/62336-67-8.png)
![2H-Cyclopenta[l]phenanthren-2-one, 1,3-bis(4-methoxyphenyl)-](http://img.cochemist.com/ccimg/62400/62336-67-8_b.png)
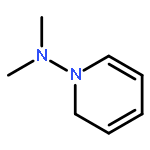

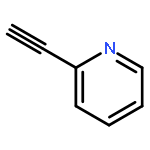
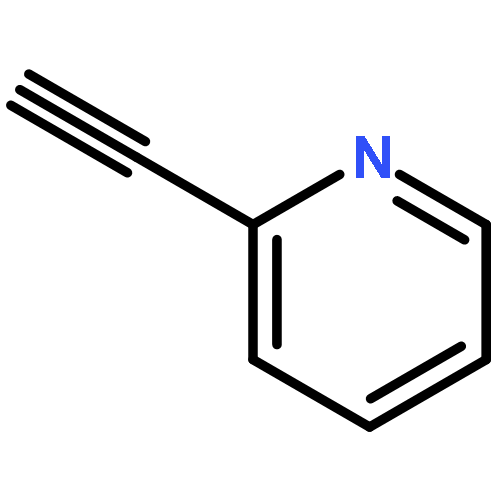
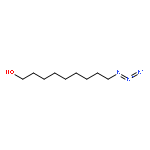
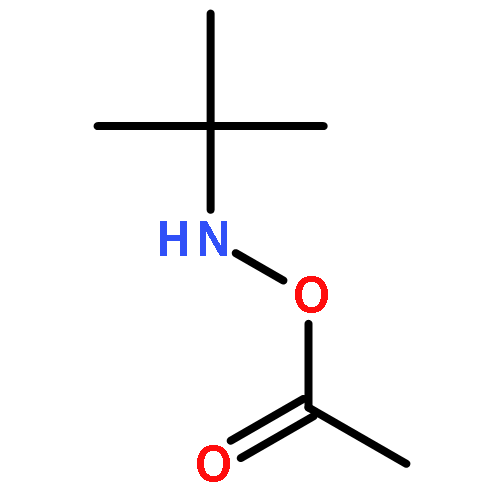
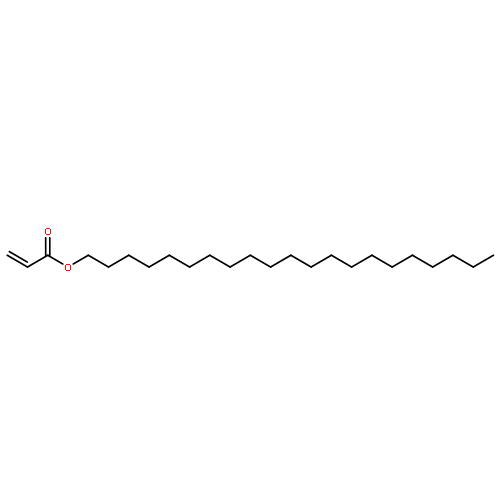


![[(E)-diazene-1,2-diyldibenzene-4,1-diyl]dimethanol](http://img.cochemist.com/ccimg/37800/37797-30-1.png)
![[(E)-diazene-1,2-diyldibenzene-4,1-diyl]dimethanol](http://img.cochemist.com/ccimg/37800/37797-30-1_b.png)

![2-[4-(DIMETHYLAMINO)PHENYL]-2-HYDROXY-1-PHENYLETHANONE](http://img.cochemist.com/ccimg/33500/33458-29-6.png)
![2-[4-(DIMETHYLAMINO)PHENYL]-2-HYDROXY-1-PHENYLETHANONE](http://img.cochemist.com/ccimg/33500/33458-29-6_b.png)
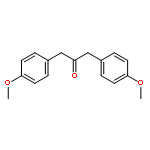
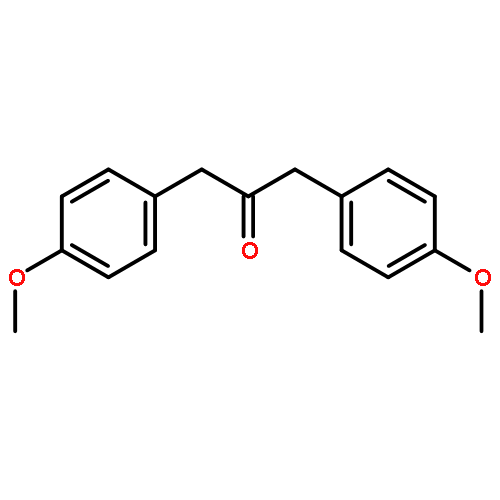
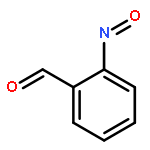
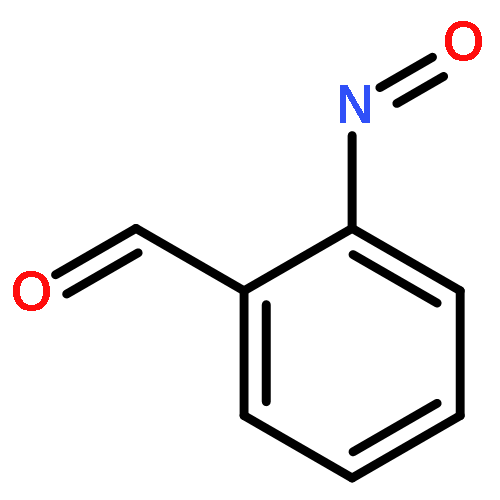

![1,1'-Biphenyl, 4,4'-bis[2-(trimethylsilyl)ethynyl]-](/data/chemimg/2248300/29619-44-1.png)
![1,1'-Biphenyl, 4,4'-bis[2-(trimethylsilyl)ethynyl]-](/data/chemimg/2248300/29619-44-1_b.png)
![Poly[oxy[(1S)-1-methyl-2-oxo-1,2-ethanediyl]]](http://img.cochemist.com/ccimg/26200/26161-42-2.png)
![Poly[oxy[(1S)-1-methyl-2-oxo-1,2-ethanediyl]]](http://img.cochemist.com/ccimg/26200/26161-42-2_b.png)
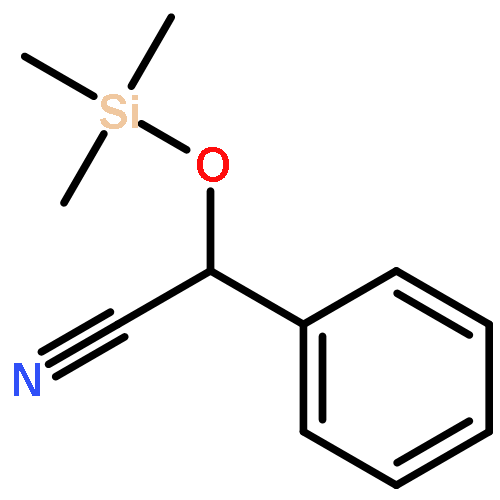
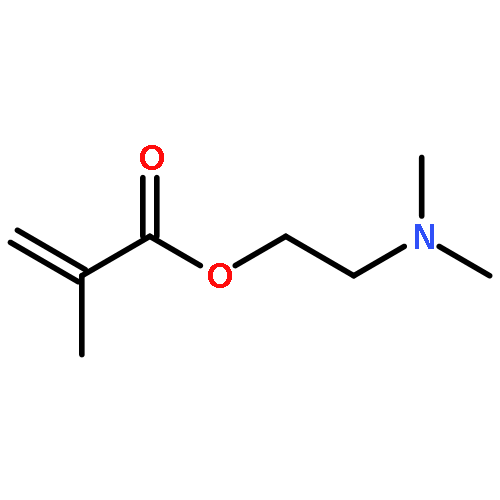
![Ethanaminium, 2-carboxy-N,N-dimethyl-N-[2-[(2-methyl-1-oxo-2-propen-1-yl)oxy]ethyl]-, inner salt](/data/chemimg/1467800/24249-95-4.png)
![Ethanaminium, 2-carboxy-N,N-dimethyl-N-[2-[(2-methyl-1-oxo-2-propen-1-yl)oxy]ethyl]-, inner salt](/data/chemimg/1467800/24249-95-4_b.png)
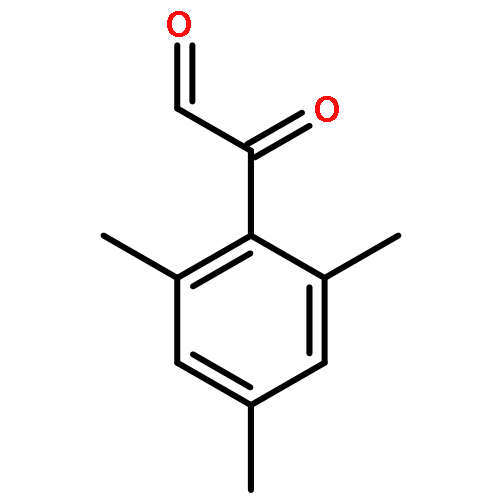
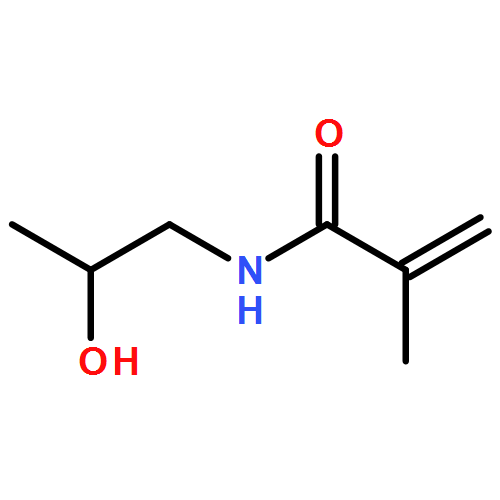

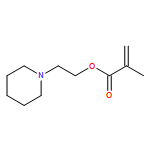
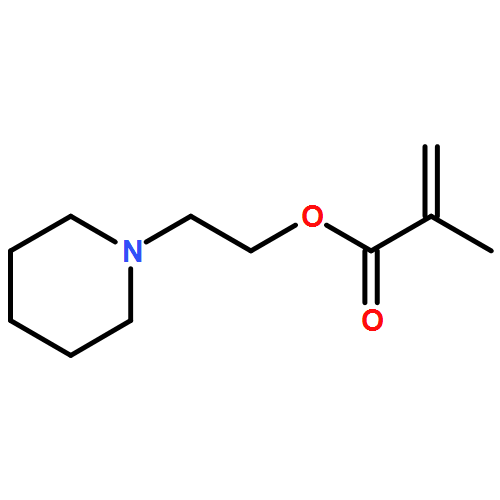
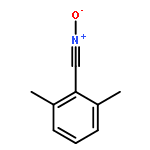
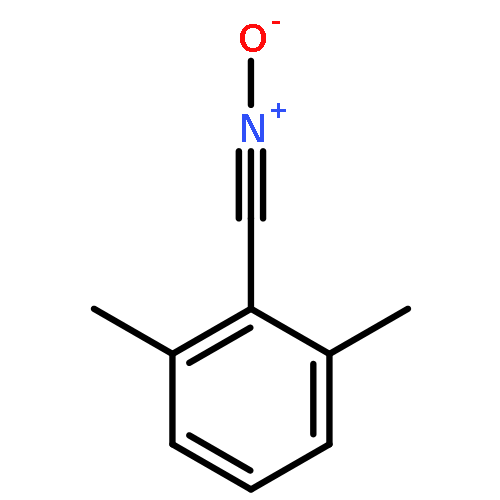
![Methanamine, N-[(4-bromophenyl)methylene]-, N-oxide](http://img.cochemist.com/ccimg/16100/16089-65-9.png)
![Methanamine, N-[(4-bromophenyl)methylene]-, N-oxide](http://img.cochemist.com/ccimg/16100/16089-65-9_b.png)
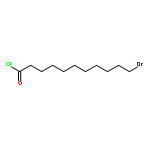
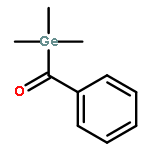
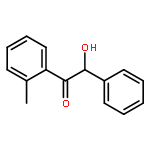
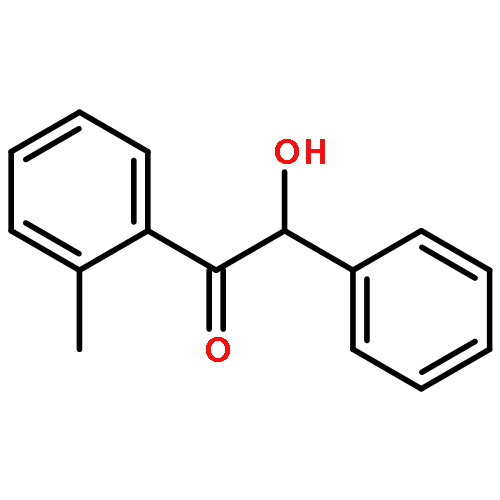
![Pyridinium, 1-[(4-ethenylphenyl)methyl]-, chloride](http://img.cochemist.com/ccimg/7600/7538-39-8.png)
![Pyridinium, 1-[(4-ethenylphenyl)methyl]-, chloride](http://img.cochemist.com/ccimg/7600/7538-39-8_b.png)
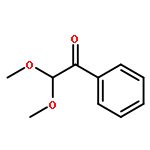
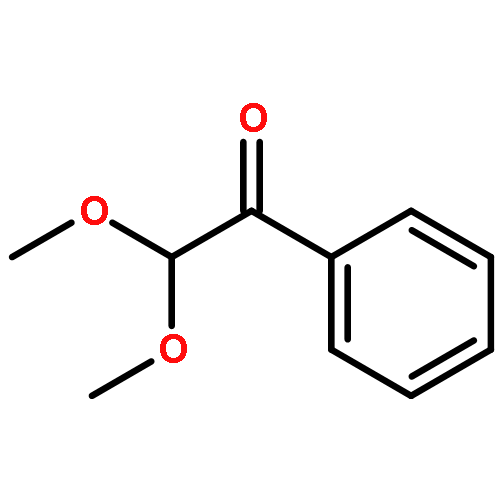
![Ethanone,1-[4-(dimethylamino)phenyl]-2-hydroxy-2-phenyl-](http://img.cochemist.com/ccimg/6400/6317-85-7.png)
![Ethanone,1-[4-(dimethylamino)phenyl]-2-hydroxy-2-phenyl-](http://img.cochemist.com/ccimg/6400/6317-85-7_b.png)


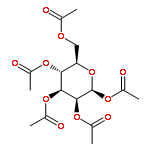
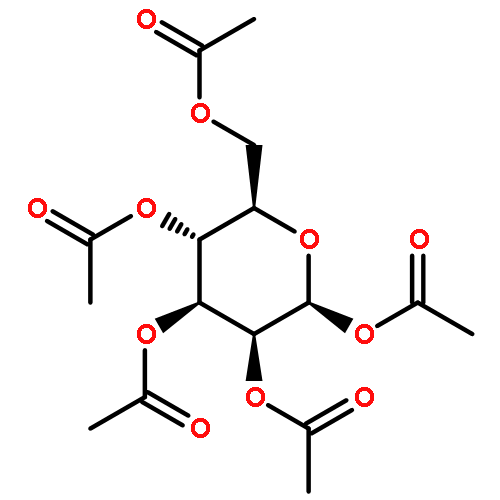
![2-Propenoic acid,2-methyl-,1,1'-[2-(hydroxymethyl)-2-[[(2-methyl-1-oxo-2-propen-1-yl)oxy]methyl]-1,3-propanediyl]ester](http://img.cochemist.com/ccimg/3600/3524-66-1.png)
![2-Propenoic acid,2-methyl-,1,1'-[2-(hydroxymethyl)-2-[[(2-methyl-1-oxo-2-propen-1-yl)oxy]methyl]-1,3-propanediyl]ester](http://img.cochemist.com/ccimg/3600/3524-66-1_b.png)
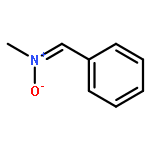
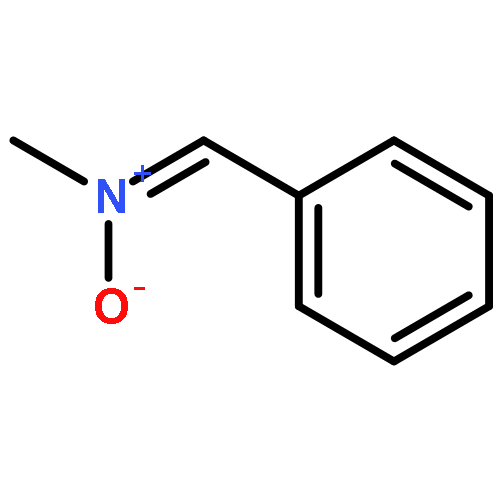

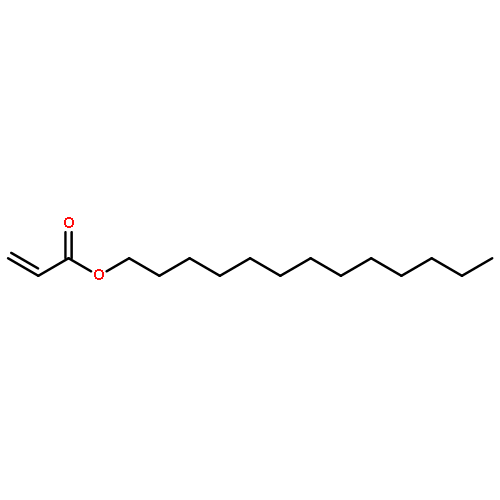
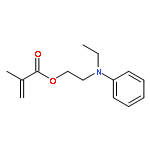
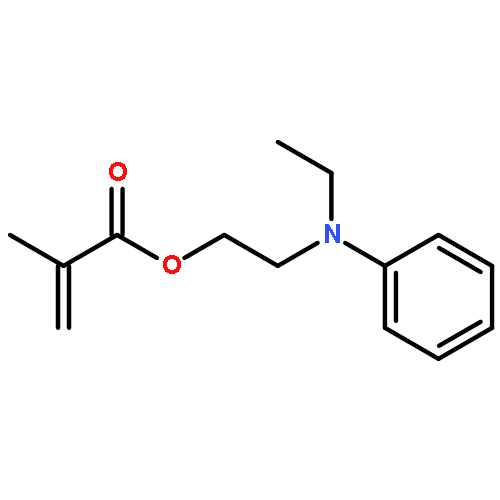
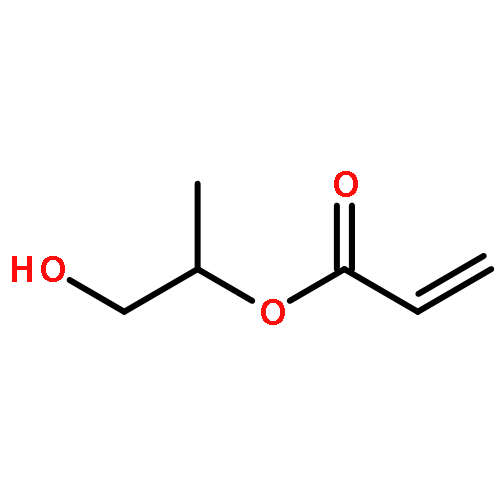

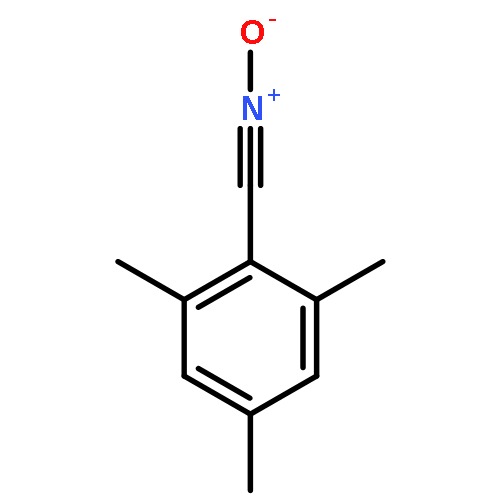
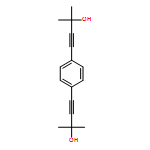
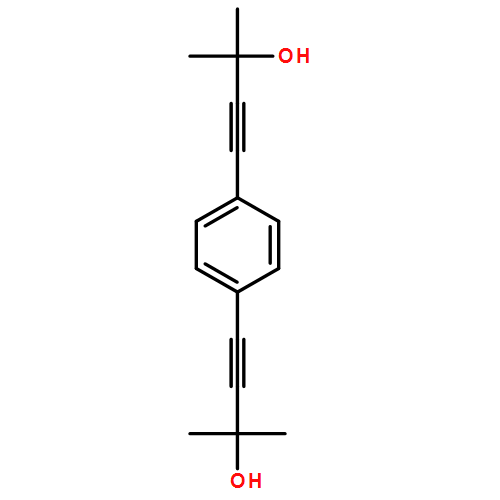
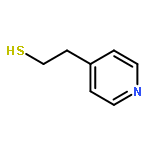

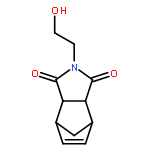
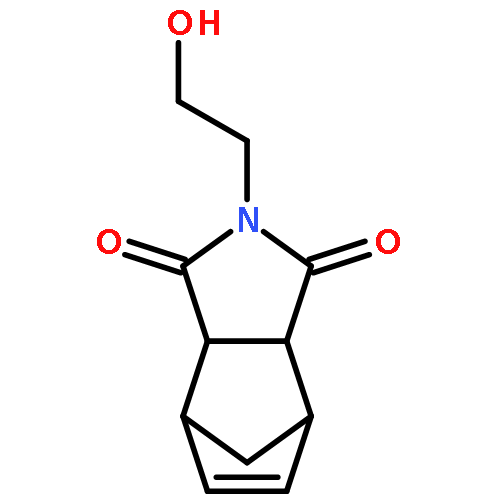
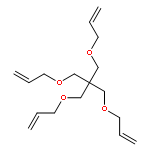




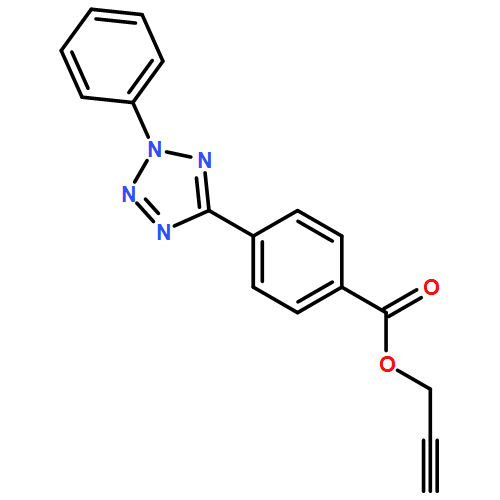
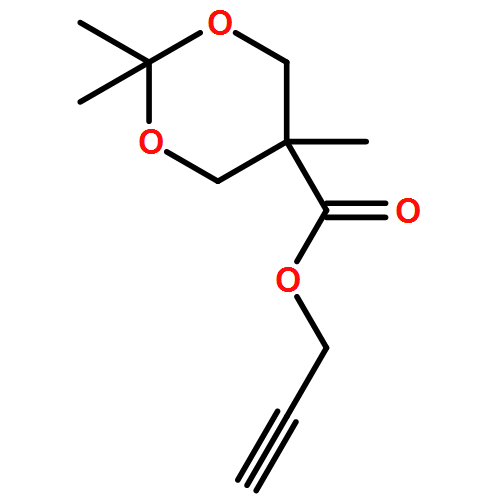
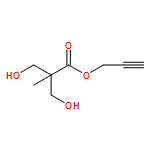
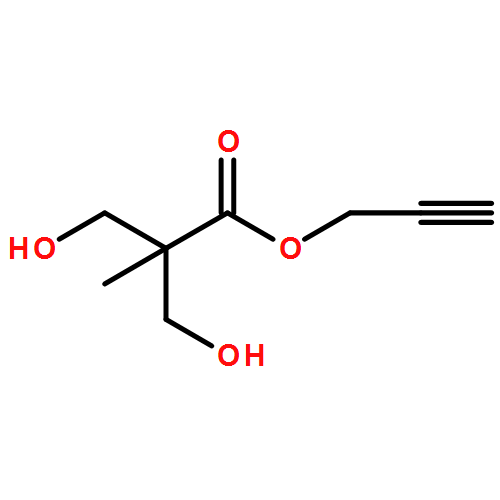
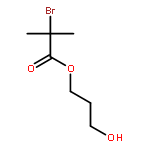
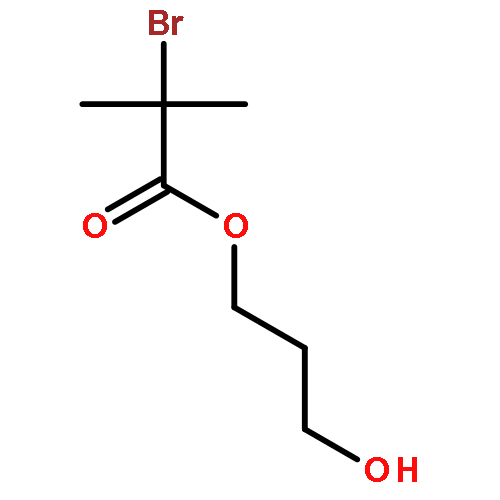
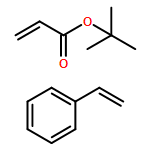
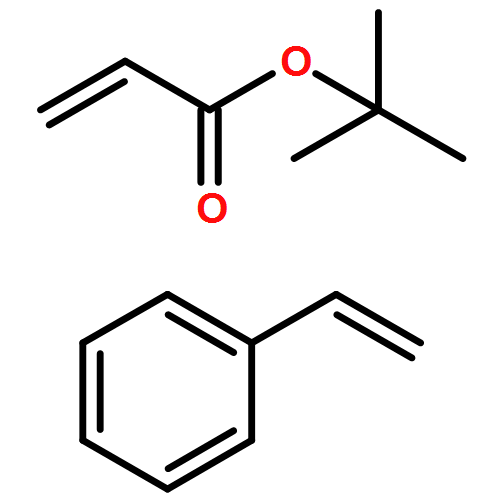
![Propanoic acid, 2,2'-[carbonothioylbis(thio)]bis[2-methyl-](/data/chemimg/104200/355120-40-0.png)
![Propanoic acid, 2,2'-[carbonothioylbis(thio)]bis[2-methyl-](/data/chemimg/104200/355120-40-0_b.png)
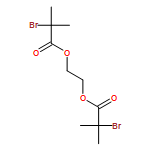
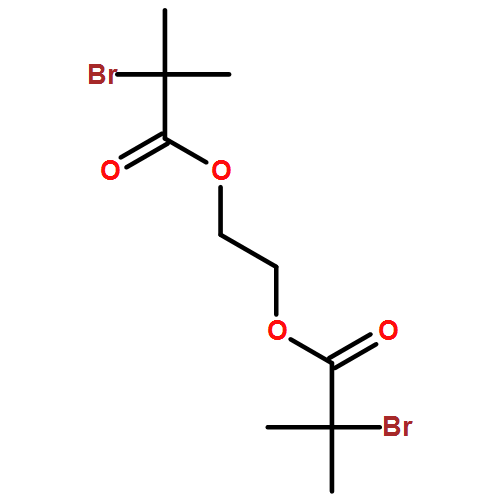


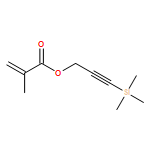
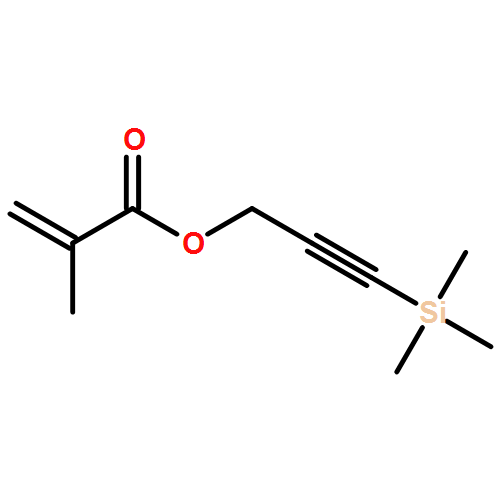
![1-Hexanol, 6-[4-(2-phenyldiazenyl)phenoxy]-](/data/chemimg/135400/162844-58-8.png)
![1-Hexanol, 6-[4-(2-phenyldiazenyl)phenoxy]-](/data/chemimg/135400/162844-58-8_b.png)




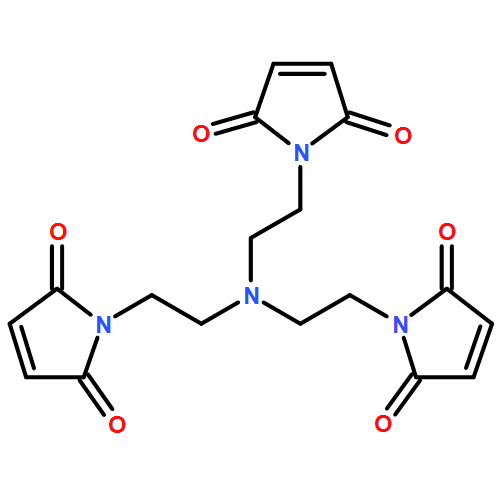
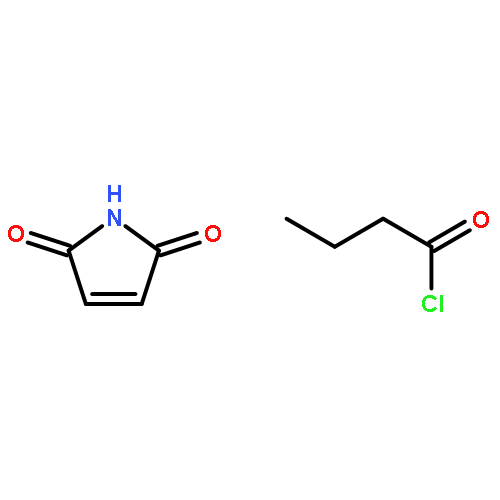
![1H-Thieno[3,4-d]imidazole-4-pentanoicacid, hexahydro-2-oxo-,2-[6-(2,5-dihydro-2,5-dioxo-1H-pyrrol-1-yl)-1-oxohexyl]hydrazide, (3aS,4S,6aR)-](http://img.cochemist.com/ccimg/117000/116919-18-7.png)
![1H-Thieno[3,4-d]imidazole-4-pentanoicacid, hexahydro-2-oxo-,2-[6-(2,5-dihydro-2,5-dioxo-1H-pyrrol-1-yl)-1-oxohexyl]hydrazide, (3aS,4S,6aR)-](http://img.cochemist.com/ccimg/117000/116919-18-7_b.png)
![Carbonotrithioic acid, bis[[4-(1,1-dimethylethyl)phenyl]methyl] ester](http://img.cochemist.com/ccimg/89700/89622-60-6.png)
![Carbonotrithioic acid, bis[[4-(1,1-dimethylethyl)phenyl]methyl] ester](http://img.cochemist.com/ccimg/89700/89622-60-6_b.png)

![2-Propenoic acid, 1-methyl-2-[[(phenylamino)carbonyl]oxy]ethyl ester](http://img.cochemist.com/ccimg/66400/66310-82-5.png)
![2-Propenoic acid, 1-methyl-2-[[(phenylamino)carbonyl]oxy]ethyl ester](http://img.cochemist.com/ccimg/66400/66310-82-5_b.png)
![2-Propenoic acid, 2-[[(phenylamino)carbonyl]oxy]ethyl ester](http://img.cochemist.com/ccimg/51800/51727-48-1.png)
![2-Propenoic acid, 2-[[(phenylamino)carbonyl]oxy]ethyl ester](http://img.cochemist.com/ccimg/51800/51727-48-1_b.png)
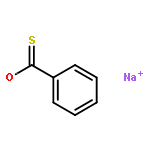

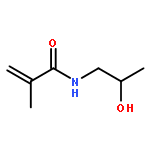
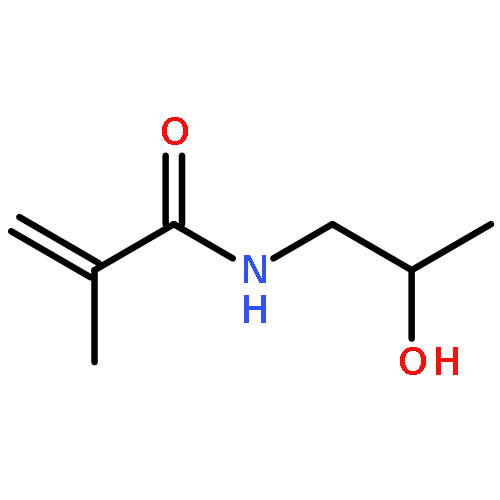
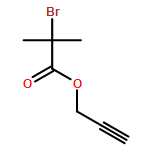
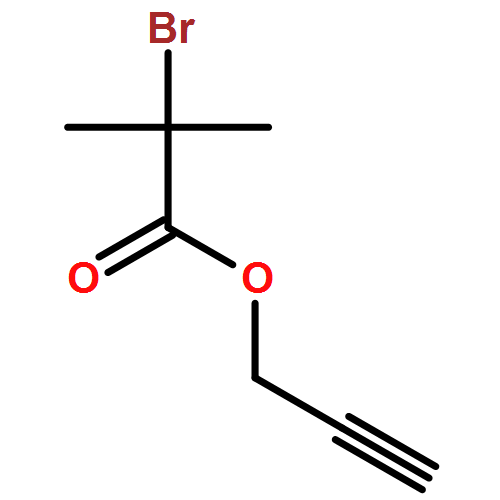

![Poly[oxy(1-oxo-1,6-hexanediyl)]](http://img.cochemist.com/ccimg/25300/25248-42-4.png)
![Poly[oxy(1-oxo-1,6-hexanediyl)]](http://img.cochemist.com/ccimg/25300/25248-42-4_b.png)


![1-[6-(2,5-dioxopyrrol-1-yl)hexyl]pyrrole-2,5-dione](http://img.cochemist.com/ccimg/4900/4856-87-5.png)
![1-[6-(2,5-dioxopyrrol-1-yl)hexyl]pyrrole-2,5-dione](http://img.cochemist.com/ccimg/4900/4856-87-5_b.png)
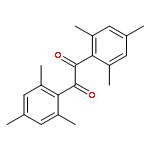
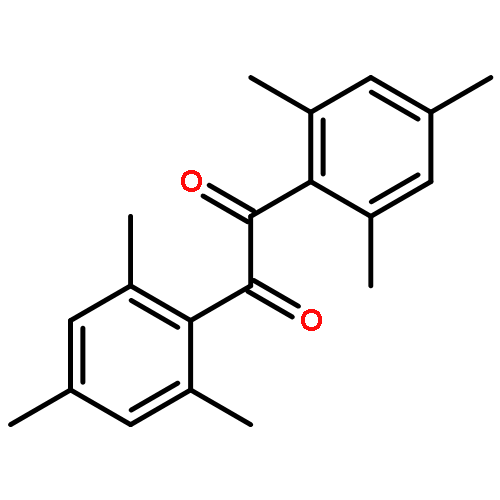
![Pyridine, 2-[(phenylsulfonyl)methyl]-](http://img.cochemist.com/ccimg/1700/1620-50-4.png)
![Pyridine, 2-[(phenylsulfonyl)methyl]-](http://img.cochemist.com/ccimg/1700/1620-50-4_b.png)









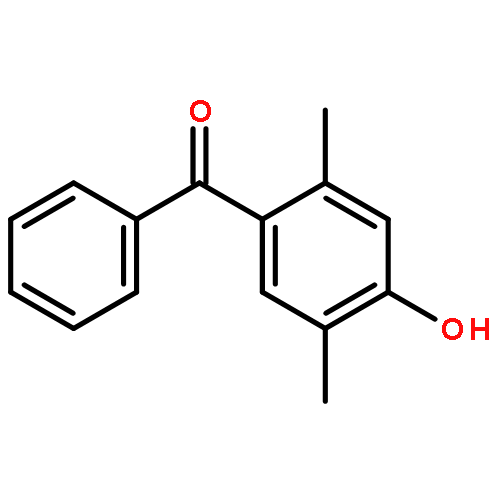



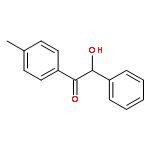
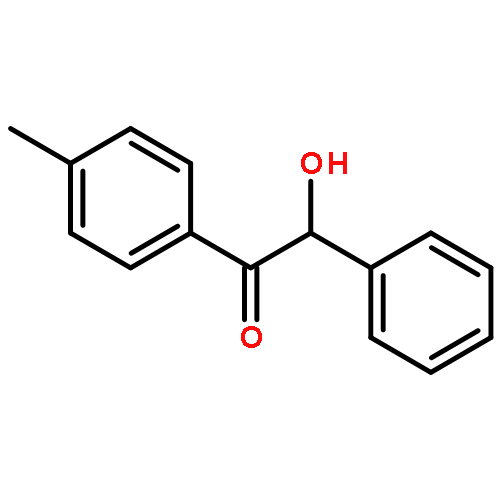


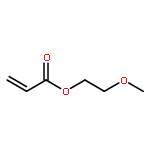
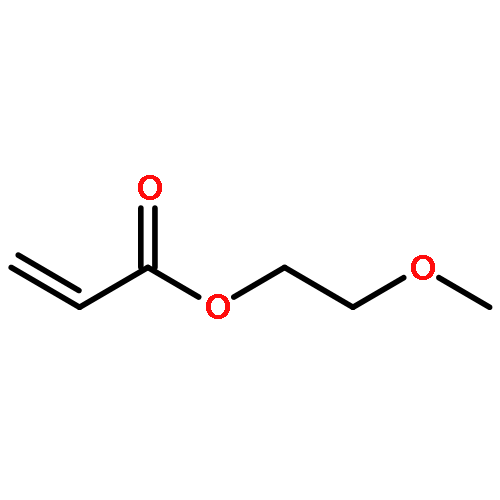
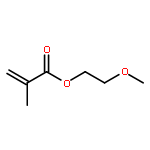
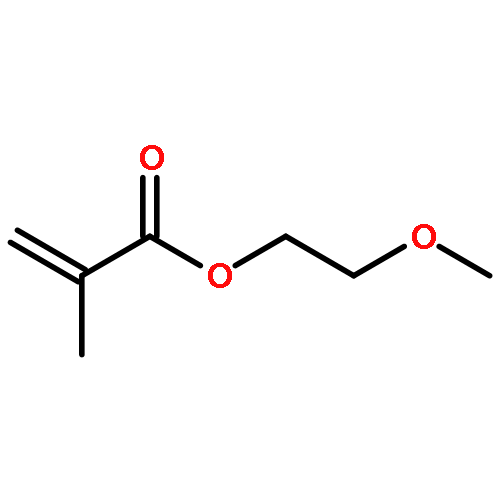
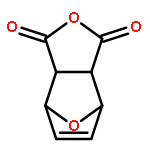
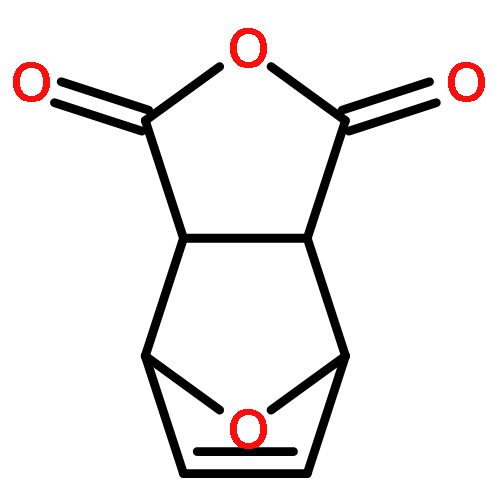
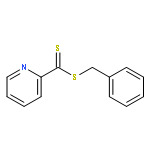
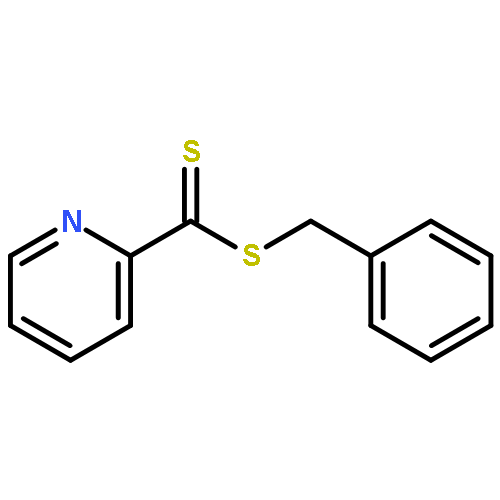
![Propanoic acid, 2-[[(ethylthio)thioxomethyl]thio]-2-methyl-](/data/chemimg/113500/881037-62-3.png)
![Propanoic acid, 2-[[(ethylthio)thioxomethyl]thio]-2-methyl-](/data/chemimg/113500/881037-62-3_b.png)
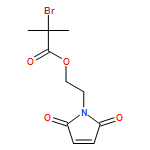
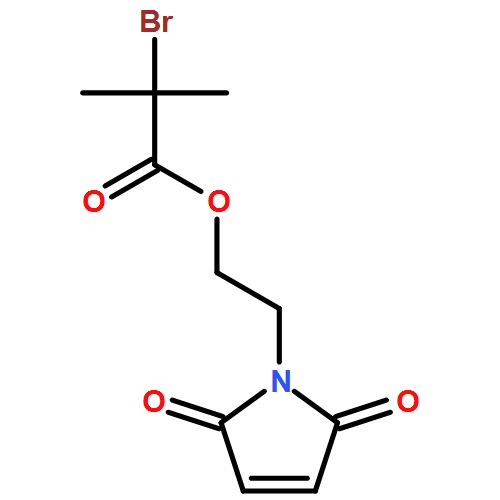



![Acetic acid,2-[(2-oxo-2-phenylethyl)thio]-](http://img.cochemist.com/ccimg/22600/22536-46-5.png)
![Acetic acid,2-[(2-oxo-2-phenylethyl)thio]-](http://img.cochemist.com/ccimg/22600/22536-46-5_b.png)