Co-reporter:Xiaoping Zhang;Xingfeng Bai;Liwen Fang
Journal of The American Society for Mass Spectrometry 2016 Volume 27( Issue 5) pp:940-943
Publication Date(Web):2016 May
DOI:10.1007/s13361-016-1339-7
In negative electrospray ionization mass spectrometry of 4-nitrobenzyl 4-hydroxybenzoates, a decarboxylation reaction, which was significantly promoted by the presence of a nitro group on the benzyl group, competed with radical elimination reactions. Density functional theory calculations indicated that decarboxylation of deprotonated 4-nitrobenzyl vanillate occurred via a radical route involving homolytic cleavage of the Cbenzyl–O bond to give a triplet ion–neutral complex, followed by decarboxylative coupling.
Co-reporter:Shanshan Wang, Cheng Guo, Ningwen Zhang, Yanqing Wu, Huarong Zhang, Kezhi Jiang
International Journal of Mass Spectrometry 2015 Volume 376() pp:6-12
Publication Date(Web):15 January 2015
DOI:10.1016/j.ijms.2014.11.003
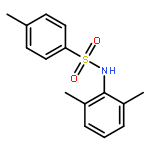
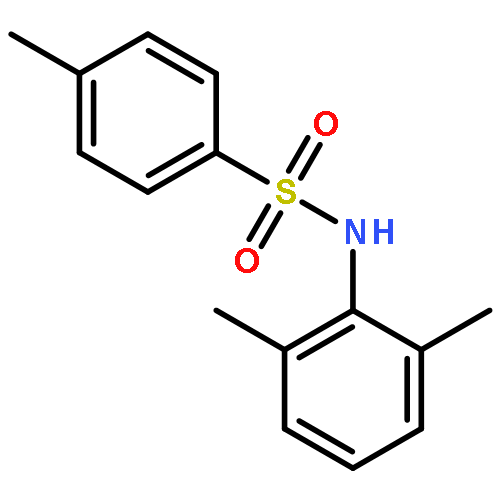
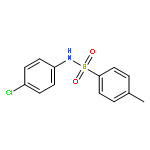
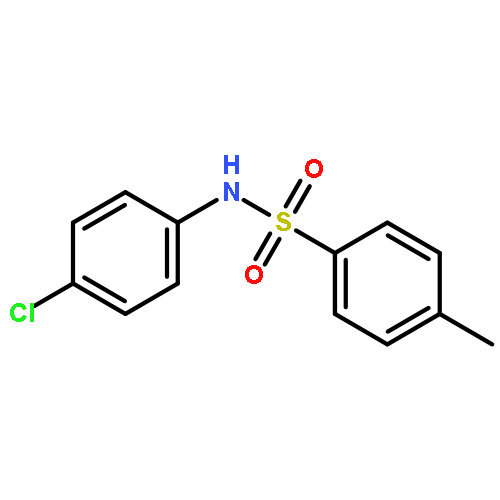
•Tosyl oxygen transfer.•INC-mediated electron transfer.•Differentiation of isomeric N-methylphenyl p-toluenesulfonamides.In this work, an interesting oxygen transfer reaction in the gas phase dissociation of N-phenyl p-toluenesulfonamides has been explored by combination of the ESI–MS techniques and theoretical calculations. The protonated molecules underwent dissociation reactions, upon collisional activation, to give the [tosyl cation/aniline] ion–neutral complex (INC), in which the coupling reactions subsequently occurred to afford an ionic species of toluenesulfinate. The subsequent reactions of the toluenesulfinate species resulted in generation of the protonated 6-iminocyclohexa- 2,4-dienone or the 2-aminophenol radical cation, via cleavage of the SO bond. The above processes involved the tosyl oxygen transfer, and calculation results indicated that both the ortho- and the para- positions at the aniline ring are favorite for the tosyl oxygen transfer, and formation of radical cation involved an INC mediated electron transfer. The isomeric N-methylphenyl p-toluenesulfonamides behaved significant difference in the CID–MS spectra, indicating that the three isomers can be distinguished by ESI–MS.
Co-reporter:Shanshan Wang;Lian Yu;Yanqing Wu
Journal of The American Society for Mass Spectrometry 2015 Volume 26( Issue 8) pp:1428-1431
Publication Date(Web):2015 August
DOI:10.1007/s13361-015-1156-4
The gas-phase dissociation chemistry of protonated N,2-diphenyl-N'-(p-toluenesulfonyl) ethanimidamides was investigated by electrospray ionization mass spectrometry in combination with density functional theory calculation. The protonated molecules underwent fragmentation via two main competing channels: (1) migration of the tosyl cation to the anilinic N atom and the subsequent loss of 2-phenylacetonitrile to afford protonated N-phenyl p-toluenesulfonamide (m/z 248); and (2) transfer of the ionizing proton to the anilinic N atom to give an ion/neutral complex of [tosyl cation / 2-phenylacetonitrile] (m/z 272) and the subsequent decomposition to yield tosyl cation (m/z 155). To the best of our knowledge, the gas-phase tosyl cation transfer has not been reported previously. For the para-substituted sulfonamides, the presence of electron-donating groups on the anilinic ring inhibits the reaction channel of the tosyl cation migration, whereas the presence of electron-withdrawing groups favors this pathway.
Co-reporter:Fei Li, Yanqing Wu, Ningwen Zhang, Jianxiong Jiang, Kezhi Jiang
International Journal of Mass Spectrometry 2014 Volume 369() pp:23-29
Publication Date(Web):15 August 2014
DOI:10.1016/j.ijms.2014.05.011
•Dissociation of the [M + H]+ ions were investigated for benzyl phenylalaninates.•Possible pathways were proposed for the competing benzyl cation transfers.•The benzyl cation transfers were supported by high resolution MS, MSn spectra, deuterium labeling experiments and theoretical calculations.•Benzyl cation is more favorable to migrate to the phenyl ring rather than to the amino N prior to the dissociation reaction of losing (H2O + CO).In this study, the competing benzyl cation transfer reactions have been explored by investigating the gas phase chemistry of the protonated benzyl phenylalaninates. Protonation at the carboxylic O atom results in the breakage of the ester CO bond to afford the benzyl cation, which undergoes the competing migration to the amino N atom or the phenyl ring C atom. Both the amino and the phenyl ring hydrogen atoms can be activated to be mobile due to the electrophilic attack of the transferring benzyl cation, and migration of the activated hydrogen atom to the carboxylic hydroxyl leads to (H2O + CO) elimination of the precursor ion. Interestingly, it is much more preferred for the benzyl cation to transfer to the phenyl ring via the amino N, leading to the stepwise benzyl cation transfer, albeit the amino N atom contains more nucleophilic affinity. The mechanistic processes have been confirmed by the MS3 spectra data, along with D-labeling experiments and theoretical calculations.
Co-reporter:Xiaoping Zhang;Fei Li;Huiqing Lv
Journal of The American Society for Mass Spectrometry 2013 Volume 24( Issue 6) pp:941-948
Publication Date(Web):2013 June
DOI:10.1007/s13361-013-0604-2
Formation of radical fragments from even-electron ions is an exception to the “even-electron rule”. In this work, ferulic acid (FA) and isoferulic acid (IFA) were used as the model compounds to probe the fragmentation mechanisms and the isomeric effects on homolytic cleavage. Elimination of methyl radical and CO2 are the two competing reactions observed in the CID-MS of [FA – H]− and [IFA – H]−, of which losing methyl radical violates the “even-electron rule”. The relative intensity of their product ions is significantly different, and thereby the two isomeric compounds can be differentiated by tandem MS. Theoretical calculations indicate that both the singlet-triplet gap and the excitation energy decrease in the transient structures, as the breaking C–O bond is lengthened. The methyl radical elimination has been rationalized as the intramolecular electronic excitation of a transient structure with an elongating C–O bond. The potential energy diagrams, completed by the addition of the energy barrier of the radical elimination, have provided a reasonable explanation of the different CID-MS behaviors of [FA – H]− and [IFA – H]−.



![[2-[(2-METHYLPHENYL)METHOXY]PHENYL]METHANOL](http://img.cochemist.com/ccimg/478200/478189-91-2.png)
![[2-[(2-METHYLPHENYL)METHOXY]PHENYL]METHANOL](http://img.cochemist.com/ccimg/478200/478189-91-2_b.png)




![Hydrazinecarbodithioic acid, [(2-chlorophenyl)methylene]-, methyl ester](http://img.cochemist.com/ccimg/146600/146539-81-3.png)
![Hydrazinecarbodithioic acid, [(2-chlorophenyl)methylene]-, methyl ester](http://img.cochemist.com/ccimg/146600/146539-81-3_b.png)
![Silane, (1,1-dimethylethyl)[(3-methylphenyl)methoxy]diphenyl-](http://img.cochemist.com/ccimg/144600/144543-13-5.png)
![Silane, (1,1-dimethylethyl)[(3-methylphenyl)methoxy]diphenyl-](http://img.cochemist.com/ccimg/144600/144543-13-5_b.png)
![Silane, [(3-methoxyphenyl)methoxy]trimethyl-](http://img.cochemist.com/ccimg/132900/132816-03-6.png)
![Silane, [(3-methoxyphenyl)methoxy]trimethyl-](http://img.cochemist.com/ccimg/132900/132816-03-6_b.png)
![Silane, trimethyl[(4-methylphenyl)methoxy]-](http://img.cochemist.com/ccimg/89300/89200-92-0.png)
![Silane, trimethyl[(4-methylphenyl)methoxy]-](http://img.cochemist.com/ccimg/89300/89200-92-0_b.png)
![L-Phenylalanine,N-[(1,1-dimethylethoxy)carbonyl]-, phenylmethyl ester](http://img.cochemist.com/ccimg/66700/66617-58-1.png)
![L-Phenylalanine,N-[(1,1-dimethylethoxy)carbonyl]-, phenylmethyl ester](http://img.cochemist.com/ccimg/66700/66617-58-1_b.png)
![HYDRAZINECARBODITHIOIC ACID, [(4-BROMOPHENYL)METHYLENE]-, METHYL ESTER](http://img.cochemist.com/ccimg/26200/26152-37-4.png)
![HYDRAZINECARBODITHIOIC ACID, [(4-BROMOPHENYL)METHYLENE]-, METHYL ESTER](http://img.cochemist.com/ccimg/26200/26152-37-4_b.png)
![Hydrazinecarbodithioic acid, [(4-chlorophenyl)methylene]-, methyl ester](http://img.cochemist.com/ccimg/26000/25996-57-0.png)
![Hydrazinecarbodithioic acid, [(4-chlorophenyl)methylene]-, methyl ester](http://img.cochemist.com/ccimg/26000/25996-57-0_b.png)