Co-reporter:Eileen G. Burke and Jennifer M. Schomaker
The Journal of Organic Chemistry September 1, 2017 Volume 82(Issue 17) pp:9038-9038
Publication Date(Web):August 10, 2017
DOI:10.1021/acs.joc.7b01506
SNO-OCTs are eight-membered heterocyclic alkynes that have fast rates of reactivity with 1,3-dipoles. In contrast to many other reported cycloalkynes, SNO-OCTs contain multiple sites for derivatization, display stability under a variety of common reaction conditions, and offer the opportunity for strain-induced ring-opening following the initial reaction of the alkyne moiety. In this paper, we describe how the unique features of SNO-OCTs can be employed to modify an oxime-bearing styrene copolymer and introduce an array of polar functionalities into the polymer. This can be achieved through both the addition of SNO-OCT to the polymer, as well as in the subsequent opening of the sulfamate ring once it has been installed in the polymer.
Co-reporter:Eileen G. Burke, Brian Gold, Trish T. Hoang, Ronald T. Raines, and Jennifer M. Schomaker
Journal of the American Chemical Society June 14, 2017 Volume 139(Issue 23) pp:8029-8029
Publication Date(Web):May 15, 2017
DOI:10.1021/jacs.7b03943
The ability to achieve predictable control over the polarization of strained cycloalkynes can influence their behavior in subsequent reactions, providing opportunities to increase both rate and chemoselectivity. A series of new heterocyclic strained cyclooctynes containing a sulfamate backbone (SNO-OCTs) were prepared under mild conditions by employing ring expansions of silylated methyleneaziridines. SNO-OCT derivative 8 outpaced even a difluorinated cyclooctyne in a 1,3-dipolar cycloaddition with benzylazide. The various orbital interactions of the propargylic and homopropargylic heteroatoms in SNO-OCT were explored both experimentally and computationally. The inclusion of these heteroatoms had a positive impact on stability and reactivity, where electronic effects could be utilized to relieve ring strain. The choice of the heteroatom combinations in various SNO-OCTs significantly affected the alkyne geometries, thus illustrating a new strategy for modulating strain via remote substituents. Additionally, this unique heteroatom activation was capable of accelerating the rate of reaction of SNO-OCT with diazoacetamide over azidoacetamide, opening the possibility of further method development in the context of chemoselective, bioorthogonal labeling.
Co-reporter:Ryan D. Reeves, Alicia M. Phelps, William A. T. Raimbach, and Jennifer M. Schomaker
Organic Letters July 7, 2017 Volume 19(Issue 13) pp:
Publication Date(Web):June 9, 2017
DOI:10.1021/acs.orglett.7b01350
Site- and regiocontrolled Au-catalyzed allene carbocyclizations furnish highly substituted cyclopentenes in >1:1 dr. Significant substitution on the substrate is tolerated, with potential to install five contiguous stereocenters after alkene functionalization. Major challenges include identifying a Au/Cu catalyst that controls both the relative rates of allene epimerization/cyclization and the facial selectivity in addition of a metal enolate to the allene. Experiments to achieve stereodivergent cyclizations and transform key cyclopentenes into useful synthetic building blocks are described.
Co-reporter:Cale Weatherly, Juliet M. Alderson, John F. Berry, Jason E. Hein, and Jennifer M. Schomaker
Organometallics April 24, 2017 Volume 36(Issue 8) pp:1649-1649
Publication Date(Web):April 3, 2017
DOI:10.1021/acs.organomet.7b00190
Catalyst-controlled, selective nitrene transfer is often challenging when both C–H and C═C bonds are present in a substrate. Interestingly, a simple change in the Ag(I):L ratio (L = bidentate N,N-donor ligand) enables tunable, chemoselective nitrene transfer that favors either C═C bond aziridination using an ∼1:1 Ag:L ratio (AgLOTf) or insertion into a C–H bond when the Ag:L ratio in the catalyst is 1:2 (AgL2OTf). In this paper, mechanistic studies, coupled with kinetic profiling of the entire reaction course, are employed to examine the reasons for this unusual behavior. Steady-state kinetics were found to be similar for both AgLOTf and AgL2OTf; both complexes yield electronically similar reactive intermediates that engage in nitrene transfer involving formation of a short-lived radical intermediate and barrierless radical recombination. Taken together, experimental and computational studies point to two effects that control tunable chemoselectivity: suppression of aziridination as the steric congestion around the silver center is increased in AgL2OTf and a decrease in the rate of C–H insertion with AgLOTf in comparison to AgL2OTf. The observation that the sterics of Ag catalysts can be varied, with minor effects on the electronic features of the putative nitrene, has important implications for the development of other silver catalysts that enable tunable, site-selective C–H bond aminations.
Co-reporter:Steven C. Schmid;Dr. Ilia A. Guzei; Jennifer M. Schomaker
Angewandte Chemie International Edition 2017 Volume 56(Issue 40) pp:12229-12233
Publication Date(Web):2017/09/25
DOI:10.1002/anie.201705202
AbstractThe reaction of rhodium-bound carbenes with strained bicyclic methylene aziridines results in a formal [3+1] ring expansion to yield highly substituted methylene azetidines with excellent regio- and stereoselectivity. The reaction appears to proceed through an ylide-type mechanism, where the unique strain and structure of the methylene aziridine promotes a ring-opening/ring-closing cascade that efficiently transfers chirality from substrate to product. The resultant products can be elaborated into new azetidine scaffolds containing vicinal tertiary-quaternary and even quaternary-quaternary stereocenters.
Co-reporter:Minsoo Ju;Dr. Cale D. Weatherly;Dr. Ilia A. Guzei; Jennifer M. Schomaker
Angewandte Chemie International Edition 2017 Volume 56(Issue 33) pp:9944-9948
Publication Date(Web):2017/08/07
DOI:10.1002/anie.201704786
AbstractAsymmetric nitrene-transfer reactions are a powerful tool for the preparation of enantioenriched amine building blocks. Reported herein are chemo- and enantioselective silver-catalyzed aminations which transform di- and trisubstituted homoallylic carbamates into [4.1.0]-carbamate-tethered aziridines in good yields and with ee values of up to 92 %. The effects of the substrate, silver counteranion, ligand, solvent, and temperature on both the chemoselectivity and ee value were explored. Stereochemical models were proposed to rationalize the observed absolute stereochemistry of the aziridines, which undergo nucleophilic ring opening to yield enantioenriched amines with no erosion in stereochemical integrity.
Co-reporter:Steven C. Schmid;Dr. Ilia A. Guzei; Jennifer M. Schomaker
Angewandte Chemie 2017 Volume 129(Issue 40) pp:12397-12401
Publication Date(Web):2017/09/25
DOI:10.1002/ange.201705202
AbstractThe reaction of rhodium-bound carbenes with strained bicyclic methylene aziridines results in a formal [3+1] ring expansion to yield highly substituted methylene azetidines with excellent regio- and stereoselectivity. The reaction appears to proceed through an ylide-type mechanism, where the unique strain and structure of the methylene aziridine promotes a ring-opening/ring-closing cascade that efficiently transfers chirality from substrate to product. The resultant products can be elaborated into new azetidine scaffolds containing vicinal tertiary-quaternary and even quaternary-quaternary stereocenters.
Co-reporter:Minsoo Ju;Dr. Cale D. Weatherly;Dr. Ilia A. Guzei; Jennifer M. Schomaker
Angewandte Chemie 2017 Volume 129(Issue 33) pp:10076-10080
Publication Date(Web):2017/08/07
DOI:10.1002/ange.201704786
AbstractAsymmetric nitrene-transfer reactions are a powerful tool for the preparation of enantioenriched amine building blocks. Reported herein are chemo- and enantioselective silver-catalyzed aminations which transform di- and trisubstituted homoallylic carbamates into [4.1.0]-carbamate-tethered aziridines in good yields and with ee values of up to 92 %. The effects of the substrate, silver counteranion, ligand, solvent, and temperature on both the chemoselectivity and ee value were explored. Stereochemical models were proposed to rationalize the observed absolute stereochemistry of the aziridines, which undergo nucleophilic ring opening to yield enantioenriched amines with no erosion in stereochemical integrity.
Co-reporter:Nicholas S. Dolan, Ryan J. Scamp, Tzuhsiung Yang, John F. Berry, and Jennifer M. Schomaker
Journal of the American Chemical Society 2016 Volume 138(Issue 44) pp:14658-14667
Publication Date(Web):October 11, 2016
DOI:10.1021/jacs.6b07981
The development of new catalysts for selective nitrene transfer is a continuing area of interest. In particular, the ability to control the chemoselectivity of intermolecular reactions in the presence of multiple reactive sites has been a long-standing challenge in the field. In this paper, we demonstrate examples of silver-catalyzed, nondirected, intermolecular nitrene transfer reactions that are both chemoselective and flexible for aziridination or C–H insertion, depending on the choice of ligand. Experimental probes present a puzzling picture of the mechanistic details of the pathways mediated by [(tBu3tpy)AgOTf]2 and (tpa)AgOTf. Computational studies elucidate these subtleties and provide guidance for the future development of new catalysts exhibiting improved tunability in group transfer reactions.
Co-reporter:Ryan J. Scamp, James G. Jirak, Nicholas S. Dolan, Ilia A. Guzei, and Jennifer M. Schomaker
Organic Letters 2016 Volume 18(Issue 12) pp:3014-3017
Publication Date(Web):May 26, 2016
DOI:10.1021/acs.orglett.6b01392
The discovery of transition metal complexes capable of promoting general, catalyst-controlled and selective carbon–hydrogen (C–H) bond amination of activated secondary C–H bonds over tertiary alkyl C(sp3)–H bonds is challenging, as substrate control often dominates when reactive nitrene intermediates are involved. In this letter, we report the design of a new silver complex, [(Py5Me2)AgOTf]2, that displays general and good-to-excellent selectivity for nitrene insertion into propargylic, benzylic, and allylic C–H bonds over tertiary alkyl C(sp3)–H bonds.
Co-reporter:Nels C. Gerstner; Christopher S. Adams; R. David Grigg; Maik Tretbar; Jared W. Rigoli
Organic Letters 2016 Volume 18(Issue 2) pp:284-287
Publication Date(Web):January 7, 2016
DOI:10.1021/acs.orglett.5b03453
Oxidative allene amination provides rapid access to densely functionalized amine-containing stereotriads through highly reactive bicyclic methyleneaziridine intermediates. This strategy has been demonstrated as a viable approach for the construction of the densely functionalized aminocyclitol core of jogyamycin, a natural product with potent antiprotozoal activity. Importantly, the flexibility of oxidative allene amination will enable the syntheses of modified aminocyclitol analogues of the jogyamycin core.
Co-reporter:Eric E. Touney, Ryan Van Hoveln, Carl T. Buttke, Michael D. Freidberg, Ilia A. Guzei, and Jennifer M. Schomaker
Organometallics 2016 Volume 35(Issue 20) pp:3436-3439
Publication Date(Web):October 10, 2016
DOI:10.1021/acs.organomet.6b00652
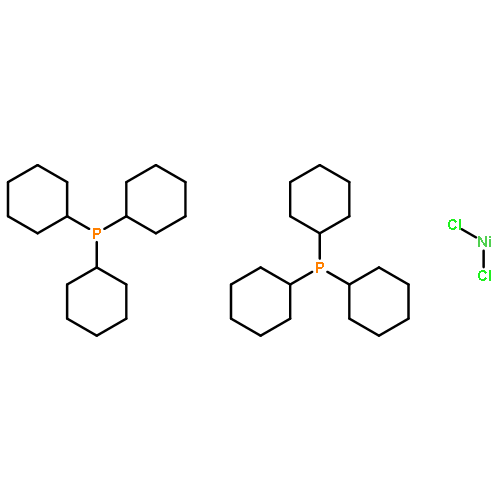
First-row transition metal catalysis offers a cheaper, more environmentally sustainable alternative to second- and third-row transition metal catalysts. Nickel has shown great promise as a tool for the borylation of unsaturated compounds to yield boronic esters, but Markovnikov-selective hydroborations of simple styrenes have not been well-explored. Herein, we report the synthesis of benzyl boronic esters via nickel-catalyzed hydroboration of styrenes using a heteroleptic N-heterocyclic carbene (NHC)–phosphine nickel complex, IMes(Cy3P)NiCl2. The IMes(Cy3P)NiCl2 complex displays a broad substrate scope and maintains the integrity of yield and regioselectivity when challenged with substrates bearing increased steric hindrance. The heteroleptic complexes also tolerate both electron-withdrawing and -donating groups, in contrast to traditional bis-phosphine and Ni(0) complexes.
Co-reporter:Alicia M. Phelps, Vincent S. Chan, José G. Napolitano, Scott W. Krabbe, Jennifer M. Schomaker, and Shashank Shekhar
The Journal of Organic Chemistry 2016 Volume 81(Issue 10) pp:4158-4169
Publication Date(Web):April 22, 2016
DOI:10.1021/acs.joc.6b00497
An iridium-catalyzed method was developed for the synthesis of imidazo-fused pyrrolopyrazines. The presence or absence of a nitrogenated ligand controlled the outcome of the reaction, leading to simple β-keto amine products in the absence of added ligand and the cyclized 7- and 8-substituted-imidazo[1,2-a]pyrrolo[2,3-e]pyrazine products in the presence of ligand. This catalyst control was conserved across a variety of ylide and amine coupling partners. The substrate was shown to act as a ligand for the iridium catalyst in the absence of other ligands via NMR spectroscopy. Kinetic studies indicated that formation of the Ir-carbene was reversible and the slow step of the reaction. These mechanistic investigations suggest that the β-keto amine products form via an intramolecular carbene N–H insertion, and the imidazopyrrolopyrazines form via an intermolecular carbene N–H insertion.
Co-reporter:Ryan Van Hoveln; Brandi M. Hudson; Henry B. Wedler; Desiree M. Bates; Gabriel Le Gros; Dean J. Tantillo
Journal of the American Chemical Society 2015 Volume 137(Issue 16) pp:5346-5354
Publication Date(Web):March 31, 2015
DOI:10.1021/ja511236d
An ongoing challenge in modern catalysis is to identify and understand new modes of reactivity promoted by earth-abundant and inexpensive first-row transition metals. Herein, we report a mechanistic study of an unusual copper(I)-catalyzed 1,3-migration of 2-bromostyrenes that reincorporates the bromine activating group into the final product with concomitant borylation of the aryl halide bond. A combination of experimental and computational studies indicated this reaction does not involve any oxidation state changes at copper; rather, migration occurs through a series of formal sigmatropic shifts. Insight provided from these studies will be used to expand the utility of aryl copper species in synthesis and develop new ligands for enantioselective copper-catalyzed halogenation.
Co-reporter:Michael F. Croisant;Ryan Van Hoveln
European Journal of Organic Chemistry 2015 Volume 2015( Issue 27) pp:5897-5907
Publication Date(Web):
DOI:10.1002/ejoc.201500561
Abstract
A thorough understanding of the mechanistic steps that occur in organometallic processes is crucial to the development of new, increasingly sophisticated transformations. This microreview discusses an unusual class of organometallic reactions that occur through formal Type I dyotropic rearrangements, defined as processes involving the interchange of two groups along a stationary scaffold by breaking and reforming two σ bonds in which at least one σ bond is a M–X bond. Examples are subdivided into two categories – stoichiometric and catalytic – and further classified by the number of atoms across which the migrating groups interchange positions. The metal can either serve as the migrating group or function as part of the stationary scaffold. Furthermore, these rearrangements can proceed through a wide variety of different mechanisms that include oxidative insertion/reductive elimination, radical processes, or pathways in which the metal does not change oxidation state.
Co-reporter:Steven C. Schmid, Ryan Van Hoveln, Jared W. Rigoli, and Jennifer M. Schomaker
Organometallics 2015 Volume 34(Issue 16) pp:4164-4173
Publication Date(Web):August 13, 2015
DOI:10.1021/acs.organomet.5b00629
A series of NHC–copper complexes was synthesized and their potential to catalyze 1,3-halogen migration explored. Increasing the steric bulk around the metal drastically improves the lifetime of NHC–CuH species and promotes 1,3-halogen migration of both 2-bromo- and 2-chlorostyrenes through transfer of an aryl halogen to a benzylic carbon with concomitant arene borylation. The NHC-based system displays a broad substrate scope with notable advantages over previously reported phosphine-based catalysts, including complete selectivity for migration versus competing benzylic borylation, increased steric tolerance, efficient aryl chloride migration, and facile formation and characterization of organocopper catalytic intermediates. Experimental evidence and DFT calculations support a mechanism proceeding through dearomatization of a benzyl copper species, followed by a 1,4-halogen shift and borylation of the resulting ArCu(I) intermediate.
Co-reporter:Eileen G. Burke ; Jennifer M. Schomaker
Angewandte Chemie International Edition 2015 Volume 54( Issue 41) pp:12097-12101
Publication Date(Web):
DOI:10.1002/anie.201504723
Abstract
Regioselectivity in the aziridination of silyl-substituted homoallenic sulfamates is readily diverted to the distal double bond of the allene to yield endocyclic bicyclic methyleneaziridines with excellent stereocontrol. Subsequent reaction with electrophilic oxygen sources initiates facile rearrangement to densely functionalized, fused azetidin-3-ones in excellent d.r., effectively transferring the axial chirality of the allene to central chirality in the products. The steric nature of the silyl group dictates which of the two rings of the fused azetidin-3-one will undergo further functionalization, providing an additional element of diversity for the preparation of enantioenriched azetidine scaffolds with potential biological activity.
Co-reporter:Eileen G. Burke ; Jennifer M. Schomaker
Angewandte Chemie 2015 Volume 127( Issue 41) pp:12265-12269
Publication Date(Web):
DOI:10.1002/ange.201504723
Abstract
Regioselectivity in the aziridination of silyl-substituted homoallenic sulfamates is readily diverted to the distal double bond of the allene to yield endocyclic bicyclic methyleneaziridines with excellent stereocontrol. Subsequent reaction with electrophilic oxygen sources initiates facile rearrangement to densely functionalized, fused azetidin-3-ones in excellent d.r., effectively transferring the axial chirality of the allene to central chirality in the products. The steric nature of the silyl group dictates which of the two rings of the fused azetidin-3-one will undergo further functionalization, providing an additional element of diversity for the preparation of enantioenriched azetidine scaffolds with potential biological activity.
Co-reporter:C. S. Adams, C. D. Weatherly, E. G. Burke and J. M. Schomaker
Chemical Society Reviews 2014 vol. 43(Issue 9) pp:3136-3163
Publication Date(Web):20 Mar 2014
DOI:10.1039/C3CS60416K
This article reviews methods for converting allenes to strained, three-membered methylene heterocycles, and also covers the reactivity of these products. Specifically, the synthesis and reactivity of methylene aziridines, allene oxides/spirodiepoxides, methylene silacyclopropanes, methylene phosphiranes, and methylene thiiranes are described, including applications to the synthesis of complex molecules. Due to the primary focus on heterocyclic motifs, the all-carbon analogue of these species (methylene cyclopropane) is only briefly discussed.
Co-reporter:Juliet M. Alderson ; Alicia M. Phelps ; Ryan J. Scamp ; Nicholas S. Dolan
Journal of the American Chemical Society 2014 Volume 136(Issue 48) pp:16720-16723
Publication Date(Web):November 11, 2014
DOI:10.1021/ja5094309
The development of readily tunable and regioselective C–H functionalization reactions that operate solely through catalyst control remains a challenge in modern organic synthesis. Herein, we report that simple silver catalysts supported by common nitrogenated ligands can be used to tune a nitrene transfer reaction between two different types of C–H bonds. The results reported herein represent the first example of ligand-controlled and site-selective silver-promoted C–H amination.
Co-reporter:R. J. Van Hoveln, S. C. Schmid, M. Tretbar, C. T. Buttke and J. M. Schomaker
Chemical Science 2014 vol. 5(Issue 12) pp:4763-4767
Publication Date(Web):04 Aug 2014
DOI:10.1039/C4SC02040E
An enantioselective Cu(I)-catalyzed 1,3-halogen migration reaction accomplishes a formal hydrobromination by transferring a bromine activating group from a sp2 carbon to a benzylic carbon in good er and with concomitant borylation of the Ar–Br bond. Computational modelling aids in understanding the reaction outcome and suggests future directions to improve the formal asymmetric hydrobromination. The benzyl bromide can be displaced with a variety of nucleophiles to produce a wide array of functionalized products.
Co-reporter:Christopher S. Adams, R. David Grigg and Jennifer M. Schomaker
Chemical Science 2014 vol. 5(Issue 8) pp:3046-3056
Publication Date(Web):13 May 2014
DOI:10.1039/C4SC01214C
Amine-containing stereotriads (‘triads’), where the nitrogen is embedded in an array of three contiguous, heteroatom-bearing chiral carbons, are key motifs in numerous bioactive natural products. Allene aziridination provides convenient access to amine triads where the position of the nitrogen and the identities of the accompanying heteroatoms can be readily manipulated. However, stereochemical flexibility, where a single allene can be selectively transformed into any possible diastereomer of a specific triad, has been elusive. Herein, we describe studies to understand how both reagent and substrate control can be effectively employed in the stereodivergent oxidative amination of allenes, with transfer of the axial chirality of an enantioenriched precursor to point chirality in each possible diastereomeric 2-amino-1,3-diol product. Application of this flexible strategy to the synthesis of all four stereoisomers of the natural product detoxinine is also presented.
Co-reporter:Jared W. Rigoli, Ilia A. Guzei, and Jennifer M. Schomaker
Organic Letters 2014 Volume 16(Issue 6) pp:1696-1699
Publication Date(Web):March 11, 2014
DOI:10.1021/ol5003576
A highly diastereoselective Ru-catalyzed oxidation/reduction sequence of bicyclic methyleneaziridines provides a facile route to complex 1-amino-2,3-diol motifs. The relative anti stereochemistry between the amine and the vicinal alcohol are proposed to result from 1,3-bischelation in the transition state by the C1 and C3 heteroatoms.
Co-reporter:R. J. Van Hoveln, S. C. Schmid and J. M. Schomaker
Organic & Biomolecular Chemistry 2014 vol. 12(Issue 39) pp:7655-7658
Publication Date(Web):13 Aug 2014
DOI:10.1039/C4OB01294A
A copper(I) catalyzed 1,3-halogen migration/borylation migrates a bromine from an sp2 carbon to a benzylic carbon with concomitant borylation of the aryl–bromine bond. This transformation proceeds via an aryl copper intermediate which can be accessed independently and then trapped with electrophiles. As such, copper-catalyzed 1,3-halogen migration provides unique and mild access to an aryl copper species that allows for rapid aromatic functionalization from an unconventional starting material.
Co-reporter:Christopher S. Adams, R. David Grigg, Jennifer M. Schomaker
Tetrahedron 2014 70(27–28) pp: 4128-4134
Publication Date(Web):
DOI:10.1016/j.tet.2014.03.084
Co-reporter:Jared W. Rigoli ; Cale D. Weatherly ; Juliet M. Alderson ; Brian T. Vo
Journal of the American Chemical Society 2013 Volume 135(Issue 46) pp:17238-17241
Publication Date(Web):November 4, 2013
DOI:10.1021/ja406654y
Organic N-containing compounds, including amines, are essential components of many biologically and pharmaceutically important molecules. One strategy for introducing nitrogen into substrates with multiple reactive bonds is to insert a monovalent N fragment (nitrene or nitrenoid) into a C–H bond or add it directly to a C═C bond. However, it has been challenging to develop well-defined catalysts capable of promoting predictable and chemoselective aminations solely through reagent control. Herein, we report remarkable chemoselective aminations that employ a single metal (Ag) and a single ligand (phenanthroline) to promote either aziridination or C–H insertion by manipulating the coordination geometry of the active catalysts.
Co-reporter:Jared W. Rigoli, Cale D. Weatherly, Brian T. Vo, Samuel Neale, Alan R. Meis, and Jennifer M. Schomaker
Organic Letters 2013 Volume 15(Issue 2) pp:290-293
Publication Date(Web):December 24, 2012
DOI:10.1021/ol303167n
Allene aziridination generates useful bicyclic methylene aziridine scaffolds that can be flexibly transformed into a range of stereochemically complex and densely functionalized amine-containing stereotriads. The scope of this chemistry has been limited by the poor chemoselectivity that often results when typical dinuclear Rh(II) catalysts are employed with homoallenic carbamates. Herein, Ag(I) catalysts that significantly improve the scope and yield of bicyclic methylene aziridines that can be prepared via allene aziridination are described.
Co-reporter:Cale D. Weatherly;Ilia A. Guzei
European Journal of Organic Chemistry 2013 Volume 2013( Issue 18) pp:3667-3670
Publication Date(Web):
DOI:10.1002/ejoc.201300416
Abstract
Nitrogen-containing stereotriads, which are defined as compounds with three adjacent stereodefined carbon atoms, are common structural motifs in several biologically relevant compounds. The 1,3-diamino-2-ol motif in particular is an important pharmacophore for which there are limited stereoselective synthetic approaches. In this communication, we describe the aminohydroxylation of a series of bicyclic methylene-aziridines obtained from the aziridination of a series of homoallenic carbamates. The unusual electronic and steric features of these useful heterocyclic scaffolds render the Os-catalyzed aminohydroxylation of the exocyclic alkene highly regio- and stereoselective. Rearrangement of the proposed N,O-aminal intermediate to a 1,3-diamino-2-one is followed by reduction with NaBH4 to deliver the desired 1,3-diamino-2-ols in good yields with high diastereomeric ratios.
Co-reporter:Alicia M. Phelps, Nicholas S. Dolan, Nathan T. Connell, Jennifer M. Schomaker
Tetrahedron 2013 69(27–28) pp: 5614-5621
Publication Date(Web):
DOI:10.1016/j.tet.2013.03.014
Co-reporter:Christopher S. Adams ; Luke A. Boralsky ; Ilia A. Guzei
Journal of the American Chemical Society 2012 Volume 134(Issue 26) pp:10807-10810
Publication Date(Web):June 18, 2012
DOI:10.1021/ja304859w
Nitrogen-containing stereotriads, compounds with three adjacent stereodefined carbons, are commonly found in biologically important molecules. However, the preparation of molecules bearing these motifs can be challenging. Herein, we describe a modular oxidation protocol which converts a substituted allene to a triply functionalized amine of the form C–X/C–N/C–Y. The key step employs a Rh-catalyzed intramolecular conversion of the allene to a strained bicyclic methylene aziridine. This reactive intermediate is further elaborated to the target products, often in one reaction vessel and with effective transfer of the axial chirality of the allene to point chirality in the stereotriad.
Co-reporter:R. David Grigg ; Ryan Van Hoveln
Journal of the American Chemical Society 2012 Volume 134(Issue 39) pp:16131-16134
Publication Date(Web):September 17, 2012
DOI:10.1021/ja306446m
A Cu(I)-catalyzed 1,3-halogen migration reaction effectively recycles an activating group by transferring bromine or iodine from a sp2 to a benzylic carbon with concomitant borylation of the Ar–X bond. The resulting benzyl halide can be reacted in the same vessel under a variety of conditions to form an additional carbon–heteroatom bond. Cross-over experiments using an isotopically enriched bromide source support intramolecular transfer of Br. The reaction is postulated to proceed via a Markovnikov hydrocupration of the o-halostyrene, oxidative addition of the resulting Cu(I) complex into the Ar–X bond, reductive elimination of the new sp3 C–X bond, and final borylation of an Ar–Cu(I) species to turn over the catalytic cycle.
Co-reporter:R. David Grigg, Jared W. Rigoli, Simon D. Pearce, and Jennifer M. Schomaker
Organic Letters 2012 Volume 14(Issue 1) pp:280-283
Publication Date(Web):December 19, 2011
DOI:10.1021/ol203055v
Propargylic amines are important intermediates for the synthesis of nitrogen-containing heterocycles. The insertion of a nitrene into a propargylic C–H bond has not been explored, despite the attention directed toward the Rh-catalyzed amination of other types of C–H bonds. In this communication, the conversion of a series of homopropargylic carbamates to propargylic carbamates and aminated allenes is described.
Co-reporter:Cale D. Weatherly, Jared W. Rigoli, and Jennifer M. Schomaker
Organic Letters 2012 Volume 14(Issue 7) pp:1704-1707
Publication Date(Web):March 20, 2012
DOI:10.1021/ol300269u
The synthesis of 1,3-diaminated stereotriads via the bis-aziridination of allenes is reported. The reactive 1,4-diazaspiro[2.2]pentane intermediates undergo a mild Brønsted acid-promoted rearrangement to yield 1,3-diaminated ketones in good yields with excellent stereocontrol. Directed reduction of the ketone can be achieved to yield a C–N/C–O/C–N stereotriad in high dr. The ability to transfer the axial chirality of the substrates to the products allows for the facile preparation of enantioenriched stereotriads from allenes in two simple steps.
Co-reporter:Jared W. Rigoli, Sara A. Moyer, Simon D. Pearce and Jennifer M. Schomaker
Organic & Biomolecular Chemistry 2012 vol. 10(Issue 9) pp:1746-1749
Publication Date(Web):22 Dec 2011
DOI:10.1039/C2OB06921K
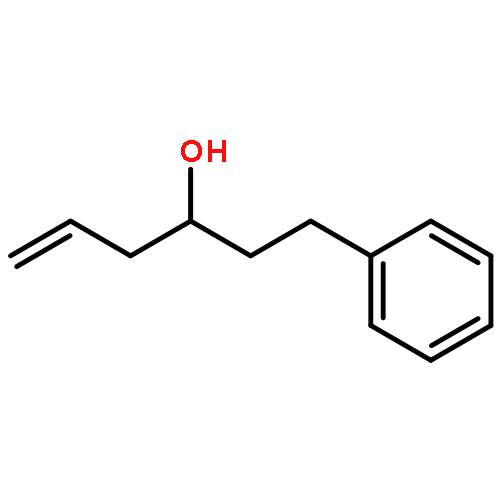
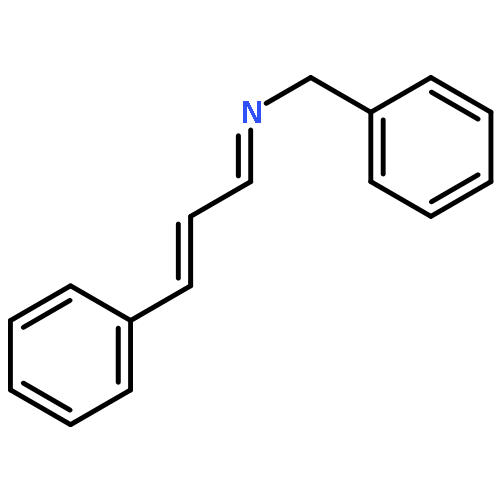
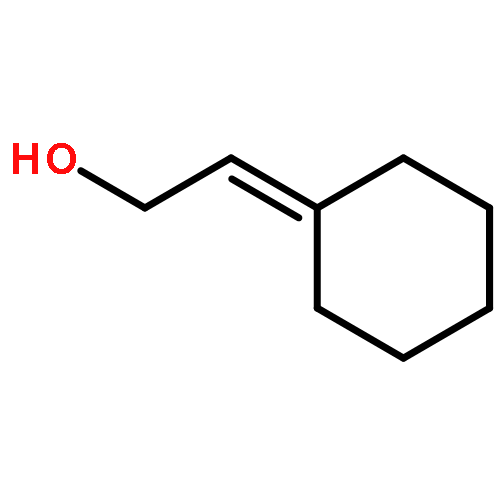
A convenient synthesis of α,β-unsaturated imines requiring only an allylic alcohol, an amine and a Ru catalyst has been developed. The use of large excesses of oxidant and the purification of sensitive intermediates can be avoided.
Co-reporter:R. David Grigg;Jared W. Rigoli;Ryan VanHoveln;Samuel Neale ; Jennifer M. Schomaker
Chemistry - A European Journal 2012 Volume 18( Issue 30) pp:9391-9396
Publication Date(Web):
DOI:10.1002/chem.201200642
Abstract
Benzylic functionalization is a convenient approach towards the conversion of readily available aromatic hydrocarbon feedstocks into more useful molecules. However, the formation of carbanionic benzyl species from benzyl halides or similar precursors is far from trivial. An alternative approach is the direct reaction of a styrene with a suitable coupling partner, but these reactions often involve the use of precious-metal transition-metal catalysts. Herein, we report the facile and convenient generation of reactive benzyl anionic species from styrenes. A CuI-catalyzed Markovnikov hydroboration of the styrenic double bond by using a bulky pinacol borane source is followed by treatment with KOtBu to facilitate a sterically induced cleavage of the CB bond to produce a benzylic carbanion. Quenching this intermediate with a variety of electrophiles, including CO2, CS2, isocyanates, and isothiocyanates, promotes CC bond formation at the benzylic carbon atom. The utility of this methodology was demonstrated in a three-step, two-pot synthesis of the nonsteroidal anti-inflammatory drug (±)-flurbiprofen.
Co-reporter:Luke A. Boralsky, Dagmara Marston, R. David Grigg, John C. Hershberger, and Jennifer M. Schomaker
Organic Letters 2011 Volume 13(Issue 8) pp:1924-1927
Publication Date(Web):March 25, 2011
DOI:10.1021/ol2002418
The oxidative functionalization of olefins is a common method for the formation of vicinal carbon−heteroatom bonds. However, oxidative methods to transform allenes into synthetic motifs containing three contiguous carbon−heteroatom bonds are much less developed. This paper describes the use of bicyclic methylene aziridines (MAs), prepared via intramolecular allene aziridination, as scaffolds for functionalization of all three allene carbons.
Co-reporter:R. David Grigg, Jennifer M. Schomaker, Vitaliy Timokhin
Tetrahedron 2011 67(24) pp: 4318-4326
Publication Date(Web):
DOI:10.1016/j.tet.2011.03.026
Co-reporter:R. J. Van Hoveln, S. C. Schmid, M. Tretbar, C. T. Buttke and J. M. Schomaker
Chemical Science (2010-Present) 2014 - vol. 5(Issue 12) pp:NaN4767-4767
Publication Date(Web):2014/08/04
DOI:10.1039/C4SC02040E
An enantioselective Cu(I)-catalyzed 1,3-halogen migration reaction accomplishes a formal hydrobromination by transferring a bromine activating group from a sp2 carbon to a benzylic carbon in good er and with concomitant borylation of the Ar–Br bond. Computational modelling aids in understanding the reaction outcome and suggests future directions to improve the formal asymmetric hydrobromination. The benzyl bromide can be displaced with a variety of nucleophiles to produce a wide array of functionalized products.
Co-reporter:C. S. Adams, C. D. Weatherly, E. G. Burke and J. M. Schomaker
Chemical Society Reviews 2014 - vol. 43(Issue 9) pp:NaN3163-3163
Publication Date(Web):2014/03/20
DOI:10.1039/C3CS60416K
This article reviews methods for converting allenes to strained, three-membered methylene heterocycles, and also covers the reactivity of these products. Specifically, the synthesis and reactivity of methylene aziridines, allene oxides/spirodiepoxides, methylene silacyclopropanes, methylene phosphiranes, and methylene thiiranes are described, including applications to the synthesis of complex molecules. Due to the primary focus on heterocyclic motifs, the all-carbon analogue of these species (methylene cyclopropane) is only briefly discussed.
Co-reporter:R. J. Van Hoveln, S. C. Schmid and J. M. Schomaker
Organic & Biomolecular Chemistry 2014 - vol. 12(Issue 39) pp:NaN7658-7658
Publication Date(Web):2014/08/13
DOI:10.1039/C4OB01294A
A copper(I) catalyzed 1,3-halogen migration/borylation migrates a bromine from an sp2 carbon to a benzylic carbon with concomitant borylation of the aryl–bromine bond. This transformation proceeds via an aryl copper intermediate which can be accessed independently and then trapped with electrophiles. As such, copper-catalyzed 1,3-halogen migration provides unique and mild access to an aryl copper species that allows for rapid aromatic functionalization from an unconventional starting material.
Co-reporter:Jared W. Rigoli, Sara A. Moyer, Simon D. Pearce and Jennifer M. Schomaker
Organic & Biomolecular Chemistry 2012 - vol. 10(Issue 9) pp:NaN1749-1749
Publication Date(Web):2011/12/22
DOI:10.1039/C2OB06921K
A convenient synthesis of α,β-unsaturated imines requiring only an allylic alcohol, an amine and a Ru catalyst has been developed. The use of large excesses of oxidant and the purification of sensitive intermediates can be avoided.
Co-reporter:Christopher S. Adams, R. David Grigg and Jennifer M. Schomaker
Chemical Science (2010-Present) 2014 - vol. 5(Issue 8) pp:NaN3056-3056
Publication Date(Web):2014/05/13
DOI:10.1039/C4SC01214C
Amine-containing stereotriads (‘triads’), where the nitrogen is embedded in an array of three contiguous, heteroatom-bearing chiral carbons, are key motifs in numerous bioactive natural products. Allene aziridination provides convenient access to amine triads where the position of the nitrogen and the identities of the accompanying heteroatoms can be readily manipulated. However, stereochemical flexibility, where a single allene can be selectively transformed into any possible diastereomer of a specific triad, has been elusive. Herein, we describe studies to understand how both reagent and substrate control can be effectively employed in the stereodivergent oxidative amination of allenes, with transfer of the axial chirality of an enantioenriched precursor to point chirality in each possible diastereomeric 2-amino-1,3-diol product. Application of this flexible strategy to the synthesis of all four stereoisomers of the natural product detoxinine is also presented.
Co-reporter:Jared W. Rigoli ; Luke A. Boralsky ; John C. Hershberger ; Dagmara Marston ; Alan R. Meis ; Ilia A. Guzei
The Journal of Organic Chemistry () pp:
Publication Date(Web):February 3, 2012
DOI:10.1021/jo3000282
Nitrogen-containing stereotriads occur in a number of biologically active compounds, but general and flexible methods to access these compounds are limited mainly to the manipulation of chiral olefins. An alternative approach is to employ a highly chemo-, regio-, and stereocontrolled allene oxidation that can install a new carbon–heteroatom bond at each of the three original allene carbons. In this paper, an intramolecular/intermolecular allene bis-aziridination is described that offers the potential to serve as a key step for the construction of stereotriads containing vicinal diaminated motifs. The resultant 1,4-diazaspiro[2.2]pentane (DASP) scaffolds contain two electronically differentiated aziridines that undergo highly regioselective ring openings at C1 with a variety of heteroatom nucleophiles to give chiral N,N-aminals. Alternatively, the same DASP intermediate can be induced to undergo a double ring-opening reaction at both C1 and C3 to yield vicinal diaminated products corresponding to formal ring opening at C3. The chirality of a propargyl alcohol is easily transferred to the DASP with good fidelity, providing a new paradigm for the construction of enantioenriched nitrogen-containing stereotriads.

![7-Azabicyclo[4.1.0]heptane,7-[S-(4-methylphenyl)-N-[(4-methylphenyl)sulfonyl]sulfonimidoyl]-](http://img.cochemist.com/ccimg/816500/816444-70-9.png)
![7-Azabicyclo[4.1.0]heptane,7-[S-(4-methylphenyl)-N-[(4-methylphenyl)sulfonyl]sulfonimidoyl]-](http://img.cochemist.com/ccimg/816500/816444-70-9_b.png)
![3,4-Nonadien-1-ol, 9-[[(1,1-dimethylethyl)diphenylsilyl]oxy]-](/data/chemimg/3722300/2015186-20-4.png)
![3,4-Nonadien-1-ol, 9-[[(1,1-dimethylethyl)diphenylsilyl]oxy]-](/data/chemimg/3722300/2015186-20-4_b.png)
![7-Azabicyclo[4.1.0]heptane-7-sulfonic acid, 2,2,2-trichloroethyl ester](http://img.cochemist.com/ccimg/917700/917616-24-1.png)
![7-Azabicyclo[4.1.0]heptane-7-sulfonic acid, 2,2,2-trichloroethyl ester](http://img.cochemist.com/ccimg/917700/917616-24-1_b.png)
![4-Pentynal, 5-[(1,1-dimethylethyl)dimethylsilyl]-](http://img.cochemist.com/ccimg/479500/479400-49-2.png)
![4-Pentynal, 5-[(1,1-dimethylethyl)dimethylsilyl]-](http://img.cochemist.com/ccimg/479500/479400-49-2_b.png)

![Benzenesulfonamide, 4-methyl-N-[S-(4-methylphenyl)sulfonimidoyl]-](http://img.cochemist.com/ccimg/94900/94892-50-9.png)
![Benzenesulfonamide, 4-methyl-N-[S-(4-methylphenyl)sulfonimidoyl]-](http://img.cochemist.com/ccimg/94900/94892-50-9_b.png)
![2-PHENYLIMIDAZO[1,2-A]QUINOLINE](http://img.cochemist.com/ccimg/75000/74944-11-9.png)
![2-PHENYLIMIDAZO[1,2-A]QUINOLINE](http://img.cochemist.com/ccimg/75000/74944-11-9_b.png)
![Benzoic acid,2-hydroxy-6-methyl-,[(1S,2R,3R,4S,5S)-5-[(3-acetylphenyl)amino]-4-amino-3-[[(dimethylamino)carbonyl]amino]-1,2-dihydroxy-3-[(1S)-1-hydroxyethyl]-2-methylcyclopentyl]methylester](http://img.cochemist.com/ccimg/23700/23668-11-3.png)
![Benzoic acid,2-hydroxy-6-methyl-,[(1S,2R,3R,4S,5S)-5-[(3-acetylphenyl)amino]-4-amino-3-[[(dimethylamino)carbonyl]amino]-1,2-dihydroxy-3-[(1S)-1-hydroxyethyl]-2-methylcyclopentyl]methylester](http://img.cochemist.com/ccimg/23700/23668-11-3_b.png)
![Imidazo[1,2-a]pyrimidine, 2-phenyl-](http://img.cochemist.com/ccimg/15800/15764-47-3.png)
![Imidazo[1,2-a]pyrimidine, 2-phenyl-](http://img.cochemist.com/ccimg/15800/15764-47-3_b.png)


![2-Phenylimidazo[1,2-a]pyridine](http://img.cochemist.com/ccimg/4200/4105-21-9.png)
![2-Phenylimidazo[1,2-a]pyridine](http://img.cochemist.com/ccimg/4200/4105-21-9_b.png)
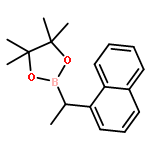
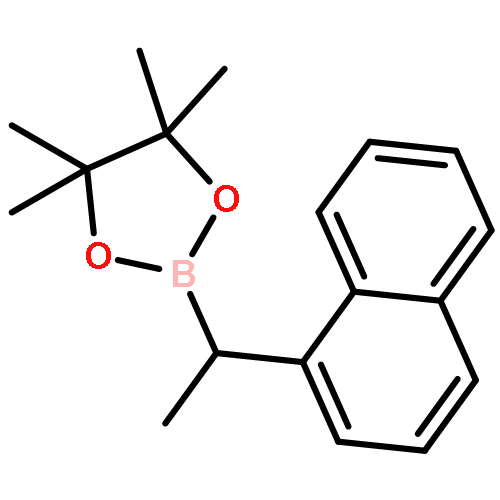
![Benzonitrile, 4-[1-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)ethyl]-](/data/chemimg/3764400/1402034-47-2.png)
![Benzonitrile, 4-[1-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)ethyl]-](/data/chemimg/3764400/1402034-47-2_b.png)
![3-Oxa-1-azabicyclo[4.1.0]heptan-2-one, 6-methyl-](http://img.cochemist.com/ccimg/918000/917908-11-3.png)
![3-Oxa-1-azabicyclo[4.1.0]heptan-2-one, 6-methyl-](http://img.cochemist.com/ccimg/918000/917908-11-3_b.png)
![3-Oxa-1-azabicyclo[4.1.0]heptan-2-one, 7-ethyl-, (6R,7R)-rel-](http://img.cochemist.com/ccimg/918000/917908-01-1.png)
![3-Oxa-1-azabicyclo[4.1.0]heptan-2-one, 7-ethyl-, (6R,7R)-rel-](http://img.cochemist.com/ccimg/918000/917908-01-1_b.png)
![Ruthenium,[6-[[bis(1,1-dimethylethyl)phosphino-kP]methylene]-N,N-diethyl-1,6-dihydro-2-pyridinemethanaminato-kN1,kN2]carbonylhydro-, (SP-5-52)-](http://img.cochemist.com/ccimg/864000/863971-63-5.png)
![Ruthenium,[6-[[bis(1,1-dimethylethyl)phosphino-kP]methylene]-N,N-diethyl-1,6-dihydro-2-pyridinemethanaminato-kN1,kN2]carbonylhydro-, (SP-5-52)-](http://img.cochemist.com/ccimg/864000/863971-63-5_b.png)
![6-[(6-BROMO-1H-[1,2,3]TRIAZOLO[4,5-B]PYRAZIN-1-YL)METHYL]QUINOLIN<WBR />E](http://img.cochemist.com/ccimg/226000/225918-07-0.png)
![6-[(6-BROMO-1H-[1,2,3]TRIAZOLO[4,5-B]PYRAZIN-1-YL)METHYL]QUINOLIN<WBR />E](http://img.cochemist.com/ccimg/226000/225918-07-0_b.png)
![[1,1-Biphenyl]-2-acetic acid, α-methyl-](/data/chemimg/3750500/126489-67-6.png)
![[1,1-Biphenyl]-2-acetic acid, α-methyl-](/data/chemimg/3750500/126489-67-6_b.png)

![Benzene, [(3-butynyloxy)methyl]-](http://img.cochemist.com/ccimg/22300/22273-77-4.png)
![Benzene, [(3-butynyloxy)methyl]-](http://img.cochemist.com/ccimg/22300/22273-77-4_b.png)
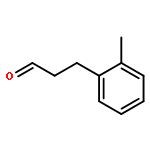
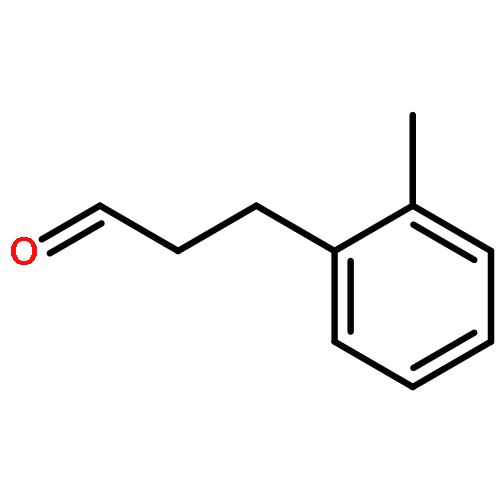
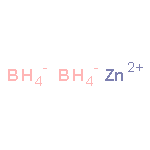
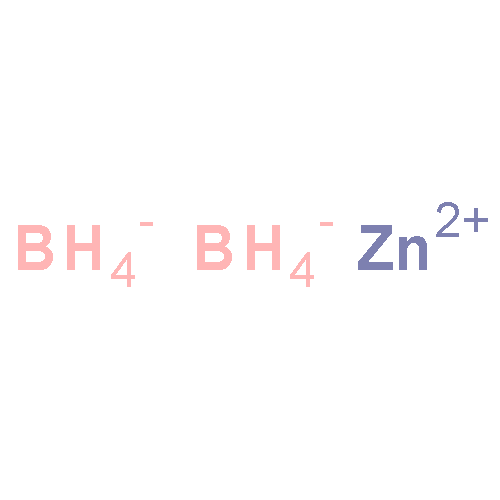
![[1,1'-Biphenyl]-4-aceticacid, a-methyl-](http://img.cochemist.com/ccimg/6400/6341-72-6.png)
![[1,1'-Biphenyl]-4-aceticacid, a-methyl-](http://img.cochemist.com/ccimg/6400/6341-72-6_b.png)
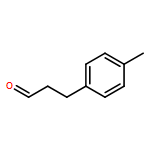
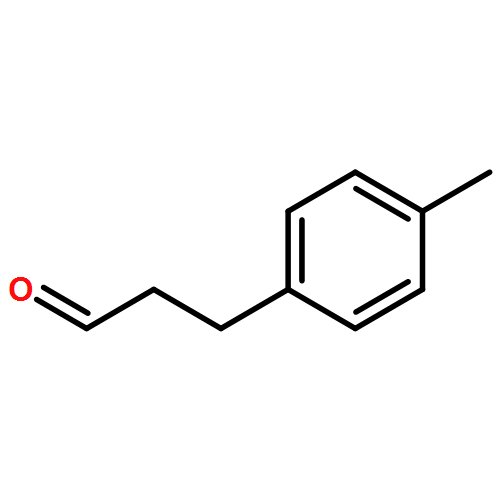
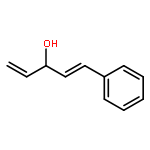
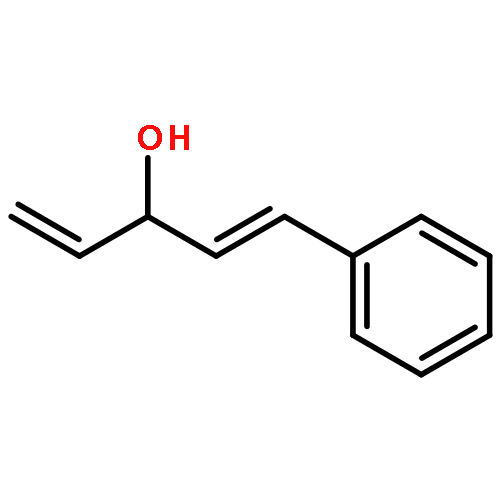



![Rhodium, bis[m-[a,a,a',a'-tetramethyl-1,3-benzenedipropanoato(2-)-kO1,kO'3:kO3,kO'1]]di-, (Rh-Rh)](http://img.cochemist.com/ccimg/819100/819050-89-0.png)
![Rhodium, bis[m-[a,a,a',a'-tetramethyl-1,3-benzenedipropanoato(2-)-kO1,kO'3:kO3,kO'1]]di-, (Rh-Rh)](http://img.cochemist.com/ccimg/819100/819050-89-0_b.png)