Co-reporter:Yongju He, Jing Li, Mengqiu Long, Shuquan Liang, Hui Xu
Journal of Non-Crystalline Solids 2017 Volume 457() pp:9-12
Publication Date(Web):1 February 2017
DOI:10.1016/j.jnoncrysol.2016.11.023
Mesoporous silica nanoparticles (MSN) with tunable pore size have been proved to be excellent potential in molecular-related applications. Herein, in this study, we designed to synthesize MSN with tunable pore size by varying the n-octadecyltrimethoxysilane (C18TMS)/tetraethylorthosilicate (TEOS) molar ratio or the ethanol (EtOH)/H2O volume ratio in a water-ethanol-ammonia system, where C18TMS acted as structure-directing agent, TEOS as silica source, ammonia as catalyst, EtOH and H2O as solvents. The as-obtained MSN were characterized by the scanning electron microscopy (SEM), transmission electron microscopy (TEM) and N2 adsorption-desorption analysis. The results showed that the as-synthesized MSN possessed spherical morphology with diameter ranging from 100 to 200 nm, and the pore size of MSN could be easily tailored from 2.4 to 6.5 nm simply by varying the C18TMS/TEOS molar ratio from 0.32 to 0.74 and the EtOH/H2O volume ratio from 3.6 to 7.5. Meanwhile, the pore volume also could be adjusted from 0.6 to 1.0 cm3/g by this strategy. These results suggested that our strategy could effectively enlarge and adjust the mesopores of the MSN. We supposed our approach may provide a platform of nanotechnology for preparation of porous silica materials with adjustable mesopores which would be great potential in practical applications where different sizes of molecules were involved.
Co-reporter:Yongju He, Shuquan Liang, Mengqiu Long, Hui Xu
Materials Science and Engineering: C 2017 Volume 78(Volume 78) pp:
Publication Date(Web):1 September 2017
DOI:10.1016/j.msec.2017.04.049
•MSN were designed as carriers for water-insoluble PTX.•The solubility of PTX was highly improved after loading into MSN.•Drug loading capacity increased as the polar parameter of solvent decreased.•Drug loading content increased as the drug/carrier mass ratio increased.•The cytotoxicity of PTX loaded MSN increased as PTX concentration increased.In this study, paclitaxel (PTX), a typical chemotherapeutic agent with poor water-solubility, was selected as the model drug to evaluate the feasibility of mesoporous silica nanoparticles (MSN) to load a hydrophobic drug in different solvents. A sol-gel method was used to synthesize MSN. Drug loading was carried out in three different solvents: dichloromethane, ethanol and dimethyl sulfoxide (DMSO) via a solvent evaporation method, and their effects on drug loading were examined. We further studied the effects of drug loading period and mass ratio of drug to carrier on drug loading capacity of MSN, as well as the in vitro drug release was analyzed. Moreover, the cytotoxic effect of PTX loaded MSN on liver carcinoma (HepG2) cells was evaluated by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. The related materials were characterized by scanning electron microscope (SEM), transmission electron microscope (TEM), dynamic light scattering (DLS), fourier transform infrared spectrometer (FTIR), small-angle x-ray scattering (SAXS), wide-angle x-ray diffraction (XRD) and N2 adsorption-desorption analyses. The results demonstrated a highly improved solubility of PTX by using MSN as drug carriers compared to free PTX. In addition, drug loading content increased as the solvent polarity parameter decreased or the drug/carrier mass ratio increased. Compared with the blank MSN, the PTX loaded MSN could produce a significant cytotoxicity on HepG2 cells. Our results indicated that MSN could be very potential drug delivery carriers for poorly water soluble drugs.Download high-res image (218KB)Download full-size image
Co-reporter:Bo Zhu, Xiaojiao Zhang, Bowen Zeng, Mingjun Li, Mengqiu Long
Organic Electronics 2017 Volume 49(Volume 49) pp:
Publication Date(Web):1 October 2017
DOI:10.1016/j.orgel.2017.06.042
•Charge mobility and half-metallicity in two dimensional metal coordination PP sheets.•All 2D metal coordination PP sheets show n-type semiconducting.•The hole mobility in all systems show anisotropy in-plane.We have investigated the electronic structures and carrier mobility of two dimensional (2D) metal coordination polyporphyrin (PP) sheets using density functional theory combined with Boltzmann transport method with relaxation time approximation. Two families of metal coordination atoms are studied: 1) 3d-transition metals (TM) Fe and Co; and 2) nonferrous metals (NM) Li and Zn. It is shown that 2H-PP, 2Li-PP and Zn-PP are direct gap semiconductors, while Fe-PP and Co-PP are spin-splitting and behave like half-metallic. Moreover, the charge mobilities have been carried out under periodic boundary conditions for infinite 2D PP systems, and found that the electron mobility is higher than that of the hole's, which indicating the n-type semiconducting characteristic. Furthermore, we also found the hole mobility in all systems show anisotropy in-plane.Download high-res image (389KB)Download full-size image
Co-reporter:Xiaojiao Zhang, Dan Zhang, Fang Xie, Xialian Zheng, Haiyan Wang, Mengqiu Long
Physics Letters A 2017 Volume 381, Issues 25–26(Issue 25) pp:
Publication Date(Web):12 July 2017
DOI:10.1016/j.physleta.2017.04.030
•The magnetic and electronic properties of Al or P doped armchair silicene nanoribbons.•The magnetic moment and band gap are dependent on the ribbon width.•The band gap oscillates with a period of three with the ribbon width increasing.Using the first-principles calculations, we investigate the geometric structure, electronic and magnetic properties of armchair silicene nanoribbons (ASiNRs) doped with aluminum (Al) or phosphorus (P) atoms. Total energy analysis shows that both Al and P atoms are preferentially doping at the edge site of ASiNRs. And the magnetism can be found in both Al and P doped systems. For Al doped ASiNRs, we find that the magnetic moment and band gap are dependent on the ribbon width. While for P doped ASiNRs, the magnetic moment always keeps 1μB and is independent of the ribbon width, meanwhile the band gap oscillates with a period of three with the ribbon width increasing. Our results present a new avenue for band engineering of SiNRs and benefit for the designing of silicone-based nano-spin-devices in nanoelectronics.
Co-reporter:Bowen Zeng, Mingjun Li, Xiaojiao Zhang, Yougen Yi, Liping Fu, and Mengqiu Long
The Journal of Physical Chemistry C 2016 Volume 120(Issue 43) pp:25037-25042
Publication Date(Web):October 10, 2016
DOI:10.1021/acs.jpcc.6b07048
Using density functional theory coupled to the Boltzmann transport equation with relaxation time approximation, we study the electronic structure and carrier mobility of graphene-like hexagonal boron phosphide (h-BP) monolayer and H-terminated armchair boron phosphide nanoribbons (ABPNRs). Our results show that the carrier mobility can reach over 104 cm2 V–1 s–1 for electron and 5 × 103 cm2 V–1 s–1 for hole in monolayer sheet. The carrier mobility in the ABPNRs is in the range of 103 to 104 cm2 V–1 s–1, and we find that the width of nanoribbon plays an important role in tuning the polarity of the carrier transport, which exhibits a distinct 3p (p is a positive integer) alternating behavior. The staggering oscillating behavior of mobility should be attributed to different bond characteristics of the edge states in the ABPNRs. Moreover, the H-terminated zigzag boron phosphide nanoribbons (ZBPNRs) have the characteristics of p-type semiconductors in electrical conduction, and the carrier mobility is increased with the width of the nanoribbons and no alternating size-dependent carrier polarity is found. The high carrier mobility and adjustable polarity of transport suggest that h-BP is a promising candidate material for application in future nanoelectronic devices.
Co-reporter:Jin Xiao
The Journal of Physical Chemistry C 2016 Volume 120(Issue 8) pp:4638-4646
Publication Date(Web):February 12, 2016
DOI:10.1021/acs.jpcc.5b12112
Blue phosphorus is a new graphene-like material which has already been proven thermostable in theory, and the synthesis of it on experiment can also be expected. Here, we have investigated the electronic structures and carrier mobilities of armchair and zigzag monolayer blue phosphorus nanoribbons (PNRs) and nanotubes (PNTs) using density functional theory combined with Boltzmann transport method with relaxation time approximation. It is found that both PNRs and PNTs are indirect-gap semiconductors with a considerable energy gap. The numerical calculation results indicate that the armchair PNTs, zigzag PNTs, and armchair PNRs have the characteristics of p-type semiconductors in electrical conduction, because the hole mobility is over 1 order larger than the electron mobility. However, the electron mobility is greater than the hole mobility in zigzag PNRs. Owing to the existing px orbitals (in-plane and along ribbon direction), which are very sensitive to the atomic structure strain, the band edges will be significantly changed under strain which results in a linear decrease of the gap of PNRs and PNTs with deformation aggravation. The charge mobilities can also be effectively regulated by the strain.
Co-reporter:Dan Zhang, Mengqiu Long, Xiaojiao Zhang and Hui Xu
RSC Advances 2015 vol. 5(Issue 117) pp:96455-96463
Publication Date(Web):26 Oct 2015
DOI:10.1039/C5RA17504F
Using the nonequilibrium Green’s function method combined with spin-polarized density functional theory, we investigate the spin-resolved electronic transport properties of devices made of poly-(terphenylene-butadiynylene) (PTB) between two symmetric ferromagnetic zigzag graphene nanoribbon (ZGNR) electrodes. The bipolar spin filtering effect, rectifying behavior, and negative differential resistance have been found. More interestingly, an on/off ratio in the order of 107 is also predicted by changing the angle between the PTB and ZGNR electrode planes. Further analyses show that the matching of the electronic wave functions among both electrodes and PTB plays a key role in the multi-functional PTB based device. And the coupling between the alkyne triple bond and the phenyl rings of PTB is critical to the value of the spin-resolved current and the on/off ratio. These phenomena suggest that the proposed PTB based devices have potential utilization in molecular spin diodes and molecular switches.
Co-reporter:Jin Xiao; Mengqiu Long; Xiaojiao Zhang; Dan Zhang; Hui Xu;Kwok Sum Chan
The Journal of Physical Chemistry Letters 2015 Volume 6(Issue 20) pp:4141-4147
Publication Date(Web):October 1, 2015
DOI:10.1021/acs.jpclett.5b01644
We have investigated the electronic structure and carrier mobility of monolayer black phosphorus nanoribbons (BPNRs) using density functional theory combined with Boltzmann transport method with relaxation time approximation. It is shown that the calculated ultrahigh electron mobility can even reach the order of 103 to 107 cm2 V–1 s–1 at room temperature. Owing to the electron mobility being higher than the hole mobility, armchair and diagonal BPNRs behave like n-type semiconductors. Comparing with the bare BPNRs, the difference between the hole and electronic mobilities can be enhanced in ribbons with the edges terminated by H atoms. Moreover, because the hole mobility is about two orders of magnitude larger than the electron mobility, zigzag BPNRs with H termination behave like p-type semiconductors. Our results indicate that BPNRs can be considered as a new kind of nanomaterial for applications in optoelectronics, nanoelectronic devices owing to the intrinsic band gap and ultrahigh charge mobility.
Co-reporter:Jun Ouyang, Mengqiu Long, Dan Zhang, Xiaojiao Zhang, Jun He, Yongli Gao
Computational Condensed Matter 2015 Volume 4() pp:40-45
Publication Date(Web):September 2015
DOI:10.1016/j.cocom.2015.08.001
Using the density functional theory (DFT) and the nonequilibrium Green's function (NEGF) method, we study the electronic structures and transport properties of zigzag boron–nitrogen–carbon nanoribbons (BNCNRs), which are constructed by the substructures of the B–N nanoribbons (BNNRs) and graphene nanoribbons (GNRs). The different position relationships (center or edge) of the BNNRs and GNRs, and the different edge patterns of the BNCNRs have been considered systematically. We found the electronic structures and transport properties of BNCNRs are significantly affected. The metallic and semiconductive properties of the BNCNRs can be modulated by the different combinations of the BNNRs and GNRs. And our results suggest BNCNRs would have potential applications in graphene-based nano-devices.
Co-reporter:Dan Zhang, Mengqiu Long, Xiaojiao Zhang, Can Cao, Hui Xu, Mingjun Li, Kowksum Chan
Chemical Physics Letters 2014 Volumes 616–617() pp:178-183
Publication Date(Web):25 November 2014
DOI:10.1016/j.cplett.2014.10.041
•The spin-dependent electronic transport properties of zigzag silicene nanoribbons.•Effects of the asymmetric edge hydrogenation on spin-dependent transport.•Bipolar spin-filtering, rectifying and giant magnetoresistance effects can be observed.Using the nonequilibrium Green's function method and the spin-polarized density functional theory, the spin-dependent electronic transport properties of zigzag silicene nanoribbons (ZSiNRs) with asymmetric edge hydrogenation have been studied. The results show that there exists nearly 100% bipolar spin-filtering behavior in the ZSiNR-based devices with antiparallel spin configuration. Moreover, rectifying behavior and giant magnetoresistance are found in the devices. Our calculation suggests ZSiNRs with asymmetric edge hydrogenation as a promising candidate material for spintronics.
Co-reporter:Shenlang Yan, Mengqiu Long, Xiaojiao Zhang, Jun He, Hui Xu, Keqiu Chen
Chemical Physics Letters 2014 Volume 608() pp:28-34
Publication Date(Web):21 July 2014
DOI:10.1016/j.cplett.2014.05.060
Highlights
- •
The spin-dependent electronic transport properties on Ni(dmit)2 device.
- •
Effects of the magnetic anchoring groups on spin-dependent transport.
- •
High spin-filtering ratios, rectification and negative differential resistance can be observed.
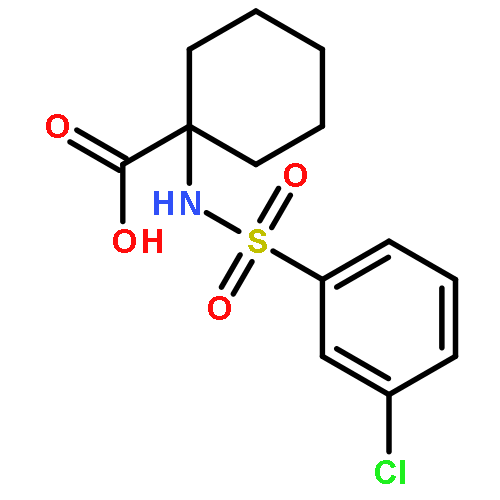
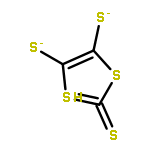
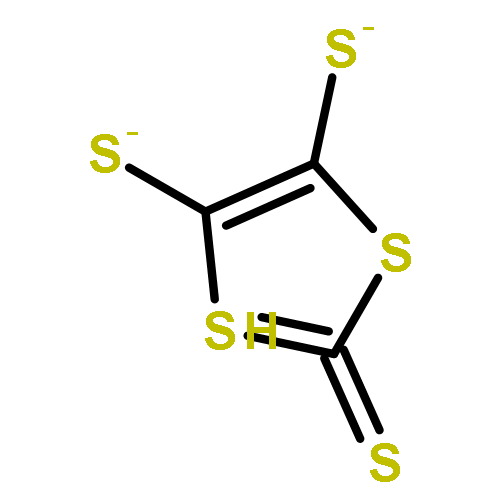
Co-reporter:Ming-Jun Li, Meng-Qiu Long, Ke-Qiu Chen, Hui Xu
Solid State Communications 2013 Volume 157() pp:62-67
Publication Date(Web):March 2013
DOI:10.1016/j.ssc.2012.12.001
The fluorination of dithiophene-tetrathiafulvalent (DT-TTF) was investigated by using the density functional theory combined with nonequilibrium Green's function method. It is demonstrated that fluorination can modify the electronic transport properties of DT-TTF. Negative differential resistance can be observed within a certain bias voltage range in 4FDT-TTF.Highlights► Fluorination effects on the electronic transport in TTF molecular junction. ► F-substitution can modify the electronic transport properties of DT-TTF. ► NDR can be observed within a certain bias voltage range in 4FDT-TTF.
Co-reporter:Mingjun Li, Dan Zhang, Yongli Gao, Can Cao, Mengqiu Long
Organic Electronics (May 2017) Volume 44() pp:168-175
Publication Date(Web):May 2017
DOI:10.1016/j.orgel.2017.02.018
Co-reporter:Ming-Jun Li, Meng-Qiu Long, Ke-Qiu Chen, Hui Xu
Solid State Communications (March 2013) Volume 157() pp:62-67
Publication Date(Web):1 March 2013
DOI:10.1016/j.ssc.2012.12.001
The fluorination of dithiophene-tetrathiafulvalent (DT-TTF) was investigated by using the density functional theory combined with nonequilibrium Green's function method. It is demonstrated that fluorination can modify the electronic transport properties of DT-TTF. Negative differential resistance can be observed within a certain bias voltage range in 4FDT-TTF.Highlights► Fluorination effects on the electronic transport in TTF molecular junction. ► F-substitution can modify the electronic transport properties of DT-TTF. ► NDR can be observed within a certain bias voltage range in 4FDT-TTF.
Co-reporter:X.Z. Wu, M.Q. Long, L.N. Chen, C. Cao, S.S. Ma, H. Xu
Physica E: Low-dimensional Systems and Nanostructures (August 2012) Volume 45() pp:82-85
Publication Date(Web):1 August 2012
DOI:10.1016/j.physe.2012.07.011
Applying nonequilibrium Green’s functions in combination with the density functional theory, we investigated the effect of amino and hydroxyl groups on the transport property of the carbon atomic chains. The results show the conductance oscillation with the length and the parity of the carbon atomic chains, and the electronic transport properties can be modulated by the sites of amino and hydroxyl groups. And also, the negative differential resistance behaviors can be observed clearly. These phenomena may originate from the interaction of side groups with the carbon atomic chains and the change in coupling degree between the molecular orbitals and electrode states.highlights► The transport property of the carbon atomic chains can be modulated by amino and hydroxyl groups. ► The conductance oscillation effect with the length and the parity of the carbon atomic chains appears in the perfect systems. ► The negative differential resistance behaviors can be observed clearly in some carbon atomic chains.